Наследственные гломерулопатии — это разнородная группа заболеваний, которые могут проявляться как нефротическим, так и нефритическим синдромом. Болезни почек нередко есть и у родственников больного, однако выяснить это удается далеко не всегда. Важную роль в понимании патогенеза этих заболеваний играет изучение молекулярных и генетических нарушений, лежащих в их основе.
Недавно французские исследователи обнаружили ген наследственного (аутосомно-рецессивного) глюкокортикоидрезистентного нефротического синдрома. Этот ген, названный NPHS2, располагается в сегменте 1q25—1q31. Он кодирует подоцин — интегральный белок клеточной мембраны подоцитов, участвующий в образовании клубочкового фильтра.
Ген наследственного (аутосомно-доминантного) фокально-сегментарного гломерулосклероза расположен на длинном плече 19-й хромосомы (сегмент 19q13). Он кодирует а-актинин-4, дефект которого делает подоциты более уязвимыми к повреждению и способствует развитию гломерулосклероза. В ряде случаев наследственного фокально-сегментарного гломерулосклероза мутации в сегменте 19q13 нет, что указывает на генетическую гетерогенность этого заболевания.
Изолированный диффузный мезангиальный склероз
Диффузный мезангиальный склероз может быть самостоятельным заболеванием. Мутации гена WT1 были найдены у четверых из десяти больных. То, что у остальных больных мутаций гена WT1 не было, говорит о генетической гетерогенности этого заболевания.
Синдром Фрайзера
Синдром Фрайзера — еще одно редкое заболевание, проявляющееся мужским псевдогермафродитизмом и почечной недостаточностью. Больные имеют нормальные женские половые органы и кариотип XY. Протеинурия возникает в детстве, затем она нарастает и приводит к развитию нефротического синдрома и ХПН в подростковом и юношеском возрасте. Фокально-сегментарный гломерулосклероз более характерен, чем диффузный мезангиальный склероз. Нефробластома возникает крайне редко. Диагноз синдрома Фрайзера может быть не установлен до тех пор, пока больную не начнут обследовать по поводу первичной аменореи. Как и при других формах дисгенезии гонад с кариотипом XY, повышен риск гонадобластом.
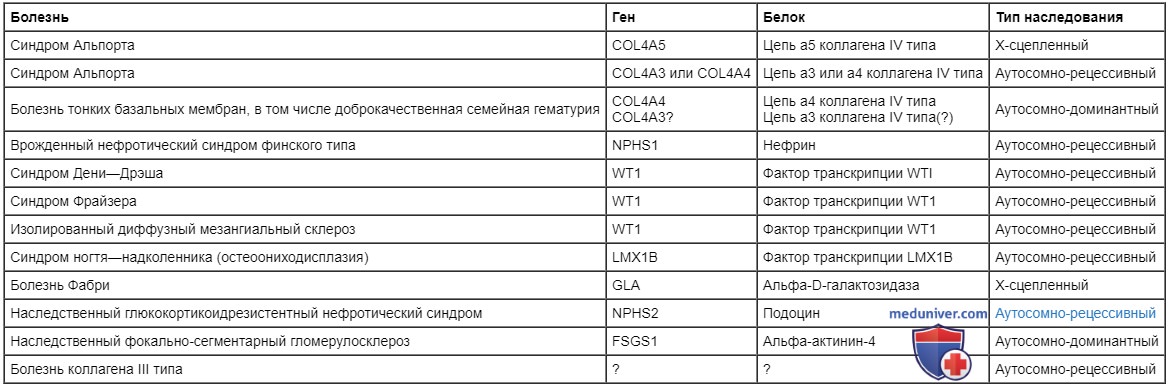
Наследственные гломерулопатии
Болезнь
Ген
Белок
Тип наследования
Синдром Альпорта
COL4A5
Цепь а5 коллагена IV типа
Х-сцепленный
Синдром Альпорта
COL4A3 или COL4A4
Цепь а3 или а4 коллагена IV типа
Аутосомно-рецессивный
Болезнь тонких базальных мембран, в том числе доброкачественная семейная гематурия
COL4A4
COL4A3?
Цепь а4 коллагена IV типа
Цепь а3 коллагена IV типа(?)