Синонимы синдрома Лобштейна. Osteopsathyrosis (Lobstein). Osteogenesis imperfecta, тип Lobstein. Идиопатический остеопсатироз. Поздний идиопатический остеопсатироз. Osteogenesis imperfecta В (Gunther). Наследственная ломкость костей (Apert). Поздняя ломкость костей. S. Ekman—Lobstein.
Определение синдрома Лобштейна. Доброкачественная форма наследственной ломкости костей. Относится к синдрому синих склер.
Авторы. Lobstein Johann Friedrich Georg Christian Martin — французский патолог и терапевт. Страсбург, 1777— 1835. Впервые синдром описали Ekman в 1788 г. и Lobstein в 1833 г.
Симптоматология синдрома Лобштейна:
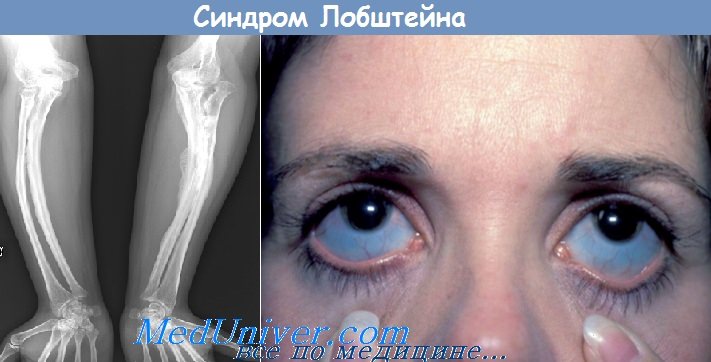
1. Патологическая ломкость костей с множественными неполными и полными переломами. Впервые переломы появляются уже после рождения (дифференциально-диагностический признак), наиболее часто в проксимальных отделах конечностей, что приводит к укорочению верхних конечностей и (относительному) удлинению нижних.
2. Нормальный рост в длину трубчатых костей (дифференциально-диагностический признак) при торможении роста в толщину диафизарных областей. Умеренный малый рост.
3. Часто — синие склеры.
4. Иногда тугоухость в результате отосклероза.
5. Биохимия крови: увеличение активности щелочной фосфатазы в сыворотке.
6. Иногда сонливость и перерастяжимость суставов с вывихами и подвывихами.
7. Рентгенологические данные: истончение компактного слоя трубчатых костей. Деформации развиваются в более позднем возрасте (искривления в области диафизов, таз в форме карточного сердца, тяжелый кифоз, сабельные голени и т. д). Нормальное развитие эпифизарных ядер окостенения.
8. Относительно доброкачественное течение: после 20-летнего возраста ломкость костей становится более умеренной.
9. Наблюдают сочетание с анетодермией и катарактой (S. Blegvad—Haxthausen).
Этиология и патогенез синдрома Лобштейна. На основании наблюдений семейного характера заболевания большинство генетиков в настоящее время придерживаются мнения об идентичности S. Lobstein и S. Vrolik (также семейного заболевания). Оба синдрома развиваются в результате мутации гена или комплекса генов.
В зависимости от характера распространения этой мутации может развиться ранняя тяжелая форма заболевания (S. Vrolik) или поздняя, доброкачественная форма (S. Lobstein). Сочетание с синими склерами или с тугоухостью может быть объяснено полифенностью гена с различной пенетрантностью и экспрессивностью или кроссинговером с функционально близким геном.
Практически во всех случаях наследование является аутосомно-доминантным; встречающийся иногда рецессивный характер наследования — генетическое правило. Последний является причиной тяжелых ранних форм. Их распознавание основывают на многообразных расстройствах периостального окостенения во всех длинных трубчатых костях в результате отсутствия активности остеобластов, в то время как процессы кальцификации протекают нормально.
Дифференциальный диагноз. S. Vrolik (см.). S. van der Hoeve (см.). S. Eddowes (см.). S. Blegvad—Haxthausen (см.). См. также Синдром синих склер.