Диффузный полипоз кишечника как наследственный синдром и предраковое заболевание
Полипозные синдромы характеризуются наличием/развитием множественных полипов в различных отделах желудочно-кишечного тракта, но часто сопровождаются другими проявлениями. Некоторые полипозные синдромы неизбежно приводят к злокачественной трансформации полипов и развитию рака (например, САТК, АСАТК); другие - не связаны с развитием рака напрямую, но могут служить индикаторами повышенного риска возникновения некоторых кишечных или внекишечных опухолей.
Семейный аденоматоз толстой кишки (САТК, АСАТК)
• Фенотип: множественные аденоматозные полипы по всей толстой кишке, периампулярные дуоденальные полипы, полипы желудка, внекишечные проявления (десмоиды и т.д.).
• Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена.
• Варианты:
- Синдром Гарднера: остеомы, десмоидные опухоли, новообразования щитовидной железы, врожденная гипертрофия пигментного эпителия сетчатки.
- Синдром Турко: опухоли головного мозга.
- Аттенуированная форма САТК (АСАТК): более поздние проявления, более проксимальное расположение полипов.
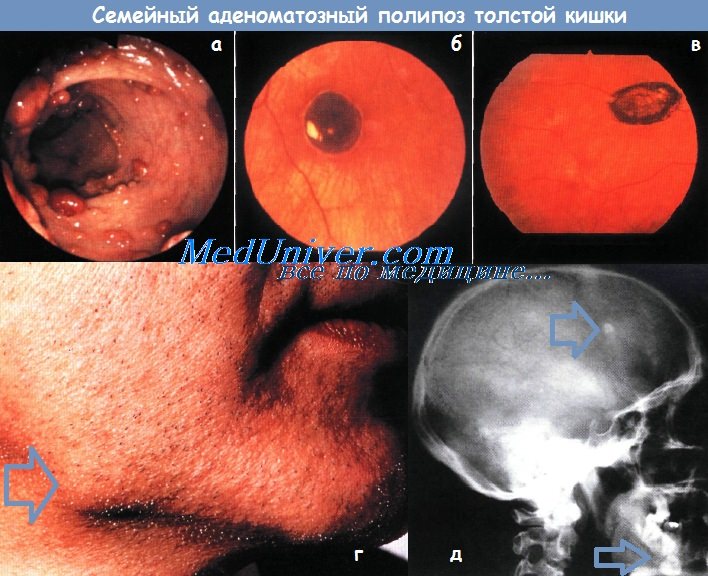
а - Колоноскопическая картина больного полипозом. Видно множество маленьких полипов.
б - Глазное дно при врожденной гипертрофии пигментного эпителия сетчатки.
У данного пациента это единственное поражение (по-липоз отсутствует).
в - Глазное дно больного с полипозом при врожденной гипертрофии пигментного эпителия сетчатки.
Одно из множественных поражений — светлый ореол с нечеткими очертаниями вокруг участка усиленной пигментации.
г - Остеома нижней челюсти у больного полипозом.
д - Рентгенограмма черепа больного полипозом. Видны остеома свода черепа и два таких же очага в нижней челюсти.
MYH-ассоциированный полипоз (МАП)
• Фенотип: часто не отличим от САТК, за исключением несколько меньшего числа полипов толстой кишки, внекишечные проявления присутствуют, но менее выражены, чем при САТК: полипы верхних отделов ЖКТ (=> периампулярный рак), остеомы, изменения зубов, наследственная гипертрофия пигментного эпителия сетчатки и др.
• Тип наследования: аутосомно-рецессивный, почти полная пенетрантность гена.
• Консультация: оба родителя и все дети являются носителями гена.
Синдром Пейтца-Егерса
• Фенотип: гамартомные полипы ЖКТ, в частности его верхних отделов, отложение меланина в коже (например, около рта, на слизистой щек и т.д.).
• Тип наследования: аутосомно-доминантный с различной пенетрантностью гена.
• Локализация гена: LKB1/STK (хромосома 19р13) и другие гены.
• Обычное течение заболевания: у большинства больных бессимптомное, в редких случаях отмечаются синдромы обструкции и кровотечения.
• Ассоциированные опухоли: умеренно повышенный риск развития опухолей ЖКТ и опухолей внекишечной локализации.
а - Полип при синдроме Пейтца-Егерса. Тотальный микроскопический препарат полипа толстой кишки.
Типичное дольчатое очертание — «голова гидры», плотная пролиферация тесно расположенных очень разветвленных желез слизистой оболочки.
Видны толстые ветвистые пучки гладких мышц в середине каждой дольки верхушки полипа.
б - Микроскопическая картина полипа при синдроме Пейтца-Егерса.
Наблюдается уплотненная и усложненная структура желез, между которыми расположена относительно бедная строма. Через верхушку полипа проходят толстые пучки гладких мышц.
Ювенильный полипоз
• Фенотип: гамартомные полипы, в 15% сочетающиеся с врожденными дефектами развития.
• Тип наследования: аутосомно-доминатный.
• Локализация гена: BMPR1A или SMAD-4 ген (хромосома 18q21) и другие гены.
• Обычное течение заболевания: средний возраст начала заболевания - 18 лет, наиболее частая локализация полипов - ректосигмоидный отдел; симптомы вариабельны: кровотечения из ЖКТ, инвагинация, выпадение прямой кишки, протеиндефицитная энтеропатия.
• Ассоциированные опухоли: значительно повышен риск развития колоректального рака.
• Диагностические критерии: > трех ювенильных полипов, полипоз всего ЖКТ, или любое количество полипов при наличии семейного анамнеза ювенильных полипов.
• Внимание: отдельные ювенильные полипы не малигнизируются.
Синдром Коудена
• Фенотип: синдром множественных гамартом из эктодермальных и в меньшей степени из эндодермальных элементов (трихолеммома — 80% случаев, макроцефалия - 40% случаев, полипоз ЖКТ - только 35% случаев, доброкачественные заболевания щитовидной и молочной желез).
• Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена.
• Локализация гена: ген опухолевой супрессии PTEN, хромосома 10q23.
• Обычное течение заболевания: симптомы заболевания появляются к 20 годам.
• Ассоциированные опухоли: риск развития опухолей ЖКТ не повышен, в 10% случаев возникает рак щитовидной железы, в 30-50% - рак молочной железы.
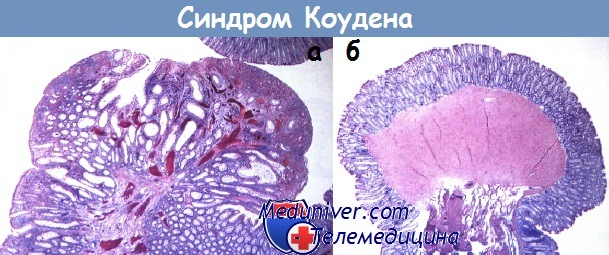
а - Синдром Коудена. Микроскопическая картина типичной гамартомы слизистой оболочки.
Пролиферация ветвистых фокально расширенных желез с умеренным количеством воспаленной стромы, которая может содержать пучки гладких мышц, фокальная эрозированная поверхность.
б - Второй тип гамартом при болезни Коудена — эквивалент лейомиомы мышечной пластинки слизистой оболочки.
• Фенотип: прогрессивный рост до/после рождения, макроцефалия, задержка умственного и психомоторного развития, другие аномалии; множественные гамартомные полипы ЖКТ; липомы; пигментные пятна на гениталиях.
• Тип наследования: аутосомно-доминантный, почти полная пенетрантность гена.
• Локализация гена: ген опухолевой супрессии PTEN, хромосома 10q23.
• Обычное течение заболевания: педиатрический аналог синдрома Коудена.
• Ассоциированные опухоли: риск развития колоректального рака, других опухолей ЖКТ и опухолей внекишечной локализации не повышен.
Синдром Кронкхайта-Канада
• Фенотип: диффузный полипоз всего ЖКТ (за исключением пищевода), эктодермальные аномалии (например, алопеция, ониходистрофия, гиперпигментация кожи).
• Тип наследования: аутосомно-доминантный.
• Локализация гена: ген опухолевой супрессии PTEN, хромосома 10q23.
• Обычное течение заболевания: диарея, белководефицитная энтеропатия, потеря веса, тошнота, рвота, анорексия, парестезии, тонико-клонические судороги, обусловленные электролитными нарушениями.
• Ассоциированные опухоли: повышенный риск развития рака желудка, толстой и прямой кишки (на момент установления диагноза отмечаются в 15% случаев).
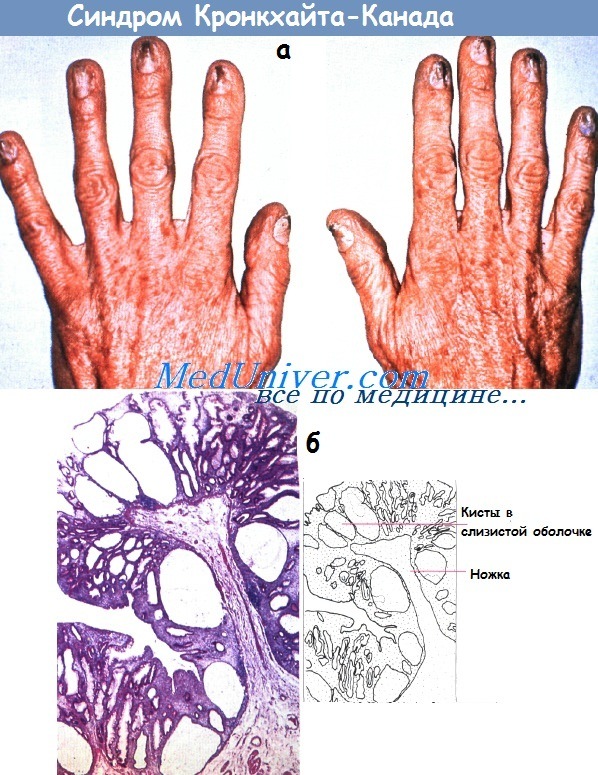
а - Ониходистрофия при синдроме Кронкхайта-Канада.
б - Гистологические проявления синдрома Кронкхайта-Канада.
Кистозные железы не диспластичны и не имеют стромальных элементов, позволяющих отличить их от других форм. Окраска гематоксилин-эозином (х 8).
Синдром гиперпластического полипоза
• Фенотип: множественные гиперпластические полипы всех отделов толстой и прямой кишки, включая крупные (> 1 см) полипы, локализованные прокси-мальнее сигмовидной кишки.
• Тип наследования: неизвестен.
• Локализация гена: неизвестна.
• Обычное течение заболевания: средний возраст 50-70 лет, специфические симптомы отсутствуют.
• Ассоциированные опухоли: повышенный риск развития колоректального рака (чаще проксимальных отделов).
Нодулярная лимфоидная гиперплазия
• Фенотип: множественные лимфоидные полипы всех отделов толстой и прямой кишки.
• Тип наследования: неизвестен.
• Локализация гена: неизвестна.
• Обычное течение заболевания: дети и взрослые, специфические симптомы отсутствуют, иногда сочетается с иммунодефицитом.
• Ассоциированные опухоли: риск развития колоректального рака не повышен.
Дополнительные исследования при полипозах кишечника
Генетическая консультация и тестирование => оценка индивидуального и семейного риска.
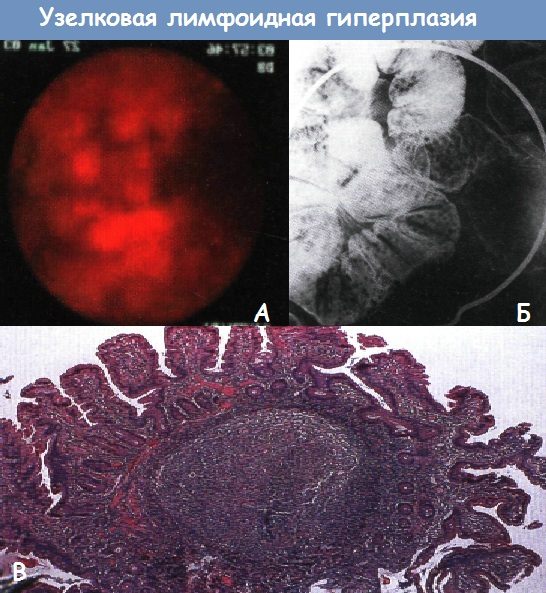
А - Капсульная эндоскопия. Лимфоидная гиперплазия у молодого пациента, неотягощенная другой патологией.
Б - Лимфоидная гиперплазия у пациента с вариабельным неклассифицируемым иммунодефицитом. Пероральная пневмоколонография. Множественные 1-2 мм рентгенопрозрачные дефекты наполнения в небольшом скоплении бария разделены нормальной гладкой слизистой оболочкой
В - Гистологический препарат подвздошной кишки с гиперплазией лимфатических узлов при иммунодефиците. Значительное увеличение пейеровых бляшек (терминальных центров) в подслизистом слое придает слизистой оболочке полиповидный вид