Синдром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) - этиология, клиника, диагностика
Синдром Корнелии де Ланге (синдром Брахмана де Ланге, синдром дегенеративного нанизма амстердамского типа) представляет собой редкое заболевание, впервые описанное Брахманом в 1916 г., но носящее имя датского педиатра де Ланге. Синдром считается очень редким, частота его составляет всего один случай на 100000 новорожденных (Beck, 1976).
а) Патогенез. Синдром Корнелии де Ланге сопровождается мутациями гена NIBPL короткого плеча пятой хромосомы (Gillis et al., 2004; Krantz et al., 2004; Tonkin et al., 2004). Недавно было выявлено, что описанные мутации является основным этиологическим фактором данного синдрома и выявляются у 27-56% пациентов (Yan et al., 2006).
Тем не менее, среди пациентов с синдромом Корнелии де Ланге выявляется ряд других хромосомных аномалий (Jackson et al., 1993). Проявления, позволяющие выявить наличие данного синдрома, отмечаются при частичной трисомии дистальной части 3-й хромосомы (3q21-3ter) и других перестройках 3-й хромосомы (DeScipio et al., 2005). Синдром дупликации 3q хромосомы (dup (3q)-синдром) только на первый взгляд имитирует синдром де Ланге (Holder et al., 1994).
Данные изменения и изменения, связывающие ген МЕСР2 с синдромом Ретта, доказывают, что следует соблюдать осторожность, рассматривая ген NIBPL как единственную причину развития синдрома де Ланге.
Синдром де Ланге может наследоваться доминантным путем; в действительности все случаи синдрома представляют собой вновь возникшие мутации. Риск повторного рождения ребенка с данной патологией составляет 2-5%.
Синдром Корнелии де Ланге:
вдавленная переносица, низкая линия роста волос, сросшиеся брови,
короткие руки и согнутые пальцы рук и ног.
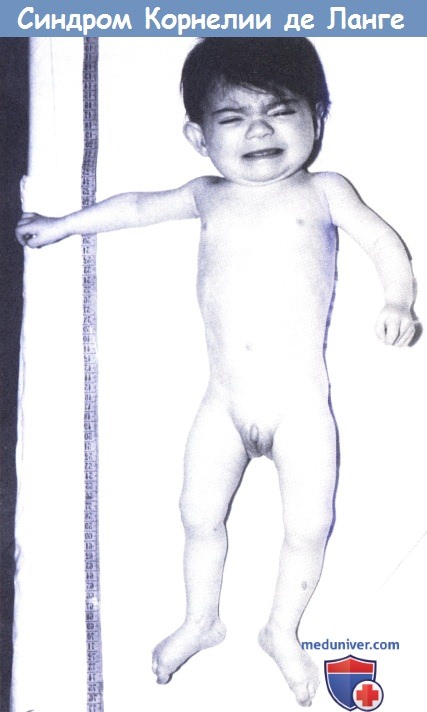
б) Клинические проявления. Синдром де Ланге является мультисистемным заболеванием, проявляющимся пре- и постнатальной задержкой роста, замедленным развитием, характерным дисморфизмом лица, мальформациями конечностей и множественными поражениями органов.
Фенотип характеризуется низкой массой при рождении, низким ростом, микроцефалией, генерализованным гирсутизмом и специфическим («любопытным») выражением лица со сросшимися бровями, низкой линией роста волос на лбу и шее, длинными ресницами, вдавленной спинкой носа, длинным подносовым желобком и вывернутыми ноздрями. Характерны плоские лопатообразные кисти и короткие конусовидные пальцы рук. Отмечается клинодактилия пятого пальца кисти.
Пальцы стоп располагаются проксимально, а возвышение большого пальца выражено незначительно. Могут встречаться более значимые аномалии конечностей, включая гипоплазию лучевой кости или уменьшение количества пальцев, часто одностороннее. Обычно отмечается микрогнатия (Hawley et al., 1985; Opitz, 1985). Зарегистрированы случаи атрофии зрительного нерва, колобомы зрительного нерва, проптоза и атрезии хоан. Часто встречается желудочно-кишечный рефлюкс, затруднения питания, пониженное слезоотделение и другие поражения глаз.
Кроме того, данное заболевание проявляется достаточно характерным поведенческим фенотипом, и в большинстве случаев серьезной или полной необучаемостью (Horsier и Oliver, 2006), часто сочетающейся с самодеструктивным поведением и избеганием социальных контактов, нередко достигая степени синдрома аутизма (Gillberg и Coleman, 2000; Arron et al., 2006; Bhuiyan et al., 2006).
Тем не менее, зарегистрированы редкие случаи пограничного или минимально нормального уровня интеллекта. По результатам работы двух исследовательских групп были выявлены значимые различия выраженности или пенетрантности некоторых фенотипов при наличии и отсутствии мутаций (Yan et al., 2006; Selicorni et al., 2007). Различные клинические проявления отмечаются и при разном характере мутаций (миссенс-мутациях и «усеченных мутациях»).
Среди пациентов с мутациями отмечается тенденция к более выраженным изменениям массы тела, роста и средней окружности головы при рождении, дисморфизму лица и нарушению речи, чем среди пациентов, у которых мутации отсутствуют.
в) Исход. В большинстве случаев тяжелые когнитивные и поведенческие нарушения сохраняются в течение всей жизни. Смертность повышена, частично вследствие нарушения функции желудочно-кишечного тракта в виде регургитации-рвоты-аспирации и развивающейся в тяжелых случаях смертельной пневмонии. Зарегистрированы отдельные случаи, когда пациенты доживали до 40 лет.