Синдром Ангельмана (ранее известный как «синдром счастливой марионетки») включает судорожные движения, беспричинный смех и задержку умственного развития различной, чаще всего серьезной умственной отсталости (Angelman 1965; Horsier и Oliver 2006). Частота в популяции составляет около 1 на 12000 живых новорожденных, с соотношением мужчин и женщин 1:1 (Steffenburg et al., 1996). Исчерпывающее описание синдрома Ангельмана в настоящее время находится в печати (Dan, 2008).
а) Патогенез. Синдром Ангельмана в большинстве случаев вызван делецией 15q11.2-12 хромосомы, сходной, но не идентичной выявляемой у детей с синдромом Прадера-Вилли (Magenis et al., 1987, Knoll et al., 1989) и наследуется по материнской линии (Knoll et al., 1989). Делеция включает ген бета-3-субъединицы ГАМК-рецептора (Saitoh et al., 1994). Среди 60-75% пациентов отмечаются делении или перестройки длинного плеча 15-й хромосомы, делеция всегда располагается на материнской хромосоме.
В небольшом количестве случаев выявляется дисомия отцовской 15-й хромосомы (Malcolm et al., 1991, Prasad и Wagstaff, 1997). Однако не менее чем у 15% пациентов выявляются нормальные хромосомы и отсутствуют признаки дисомии. В некоторых случаях такого рода возможно повторное рождение больных детей среди родственников (Clayton-Smith, 1992). Такие случаи могут быть связаны с доминантной мутацией гена UBE3A (Kishino et al., 1997) на 15q11-13 хромосоме, приводящей к возникновению фенотипа Ангельмана только при передаче по женской линии (Wagstaff et al., 1993).
Различия клинических проявлений зависят от происхождения генетического дефекта (Lossie et al., 2001). Вызванные делециями формы, обычно имеют более выраженный характер, чем формы, вызванные единичной мутацией гена или другими генетическими дефектами. Большинство случаев заболевания носит изолированный характер, но зарегистрированы и семейные случаи.
б) Диагностика. Диагноз подтверждается в случае, если клинический фенотип соответствует положительному FISH-тесту. У младенцев (van Lierde et al., 1990, Yamada и Volpe, 1990) даже при использовании предложенных критериев диагноз зачастую затруднен (Williams et al., 1995). Весьма вероятным диагноз представляется при сочетании задержки умственного развития с проявлениями аутизма, жизнерадостным поведением, атаксией и эпилептическими приступами (часто «минимально» выраженными).
На основании апраксической походки, стереотипиях движений рук и (в некоторых случаях) гипервентиляции возможна ошибочная постановка диагноза синдрома Ретта у девочек. Тем не менее, при раннем (до года) начале припадков, жизнерадостном настроении и дисморфизме вероятность неверной диагностики можно исключить.
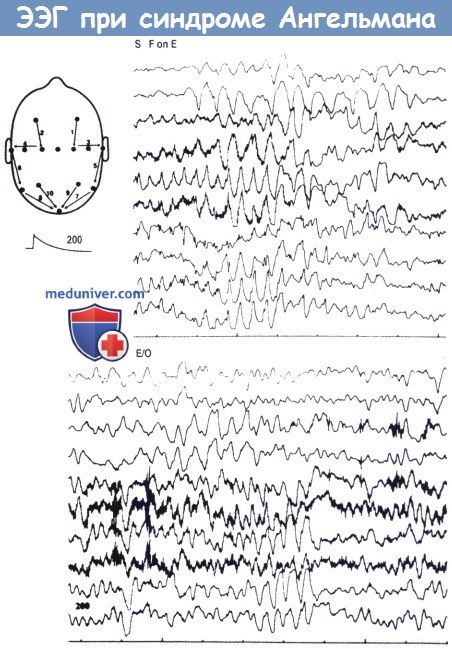
Типичные изменения на ЭЭГ двух детей различного возраста (верхний рисунок —семь лет, нижний рисунок — два года), страдающих синдромом Ангельмана.
Следует обратить внимание на калибровку. В обоих случаях отмечается повышенная ритмичная тета-активность, чередующаяся с эпизодами медленного ритма, в частности, в области передней половины двух полушарий.
На верхнем рисунке эпизоды активности с частотой 3-4/с, чередующиеся со слабовыраженными острыми волнами, отмечаются в задней трети головы в случае, когда помощник закрывает пациенту глаза.
S — закрыты, F on Е — «пальцы закрывают глаза», Е/О — глаза открыты.
в) Клинические проявления. Клинические проявления включают серьезную умственную отсталость с тяжелым нарушением речи, атаксию и заметные спастические нарушения движений верхних конечностей, а иногда и туловища; данные двигательные нарушения являются особым типом миоклонуса (Guerrini et al., 1996; Beckung et al., 2004). Немотивированные приступы смеха являются характерным, но не основным проявлением, в то время как жизнерадостное настроение является постоянным признаком (Williams и Frias, 1982).
У 86% больных детей отмечаются приступы, которые часто имеют повторный характер, но длятся недолго и чаще всего являются не тонико-клоническими припадками, а атипичными абсансами или тоническими или атоническими припадками (Dorries et al., 1988; Viani et al., 1995; Laan et al., 1997). На ЭЭГ часто встречаются характерные изменения в виде эпизодов медленных (3 Гц) волн, особенно в задних отделах, часто зазубренных и в отдельных случаях связанных с истинными пиками (Boyd et al, 1988).
На КТ и МРТ каких-либо специфичных изменений не выявляется, но возможно наличие небольшого расширения желудочков и/или околомозгового пространства (Dorries et al., 1988).
Способность к передвижению появляется поздно, часто после 5-6 лет; походка имеет аномальный характер с широко расставленными ногами и атаксическими и апраксическими признаками (Sugimoto et al., 1992).
Дисморфический синдром характеризуется умеренной выраженностью и может быть незаметен в детском возрасте. Прогнатизм обычно развивается только в возрасте нескольких лет.
Большинство случаев заболевания носит изолированный характер, но зарегистрированы и семейные случаи.
Только недавно были опубликованы результаты исследований, подтверждающих мнение о специфическом поведенческом фенотипе данного синдрома, и еще слишком рано делать предположения в данном направлении. Результаты одного из исследований взрослых и молодых пациентов, страдающих синдромом Ангельмана (Clayton-Smith, 1993), свидетельствуют о том, что гиперактивность, часто наиболее выраженная в раннем детском возрасте, в позднем детском возрасте обычно сменяется более контролируемым поведением.
Результаты большинства исследований в настоящее время позволяют предположить, что мутизм может быть постоянным проявлением, но некоторые пациенты могут освоить язык жестов. В одном из центров наблюдался мальчик с сочетанием синдрома Ангельмана и аутизма. У многих детей с синдромом Ангельмана отмечаются заметные аутистические проявления, такие как упрямство, настаивание на одном и том же и увлеченность водой, и в то же время они кажутся счастливыми и общительными, так как им нравится контактировать с другими людьми. Тем не менее, контакт осуществим лишь при выполнении их собственных условий.
г) Исход. Прогноз неблагоприятный, отмечается серьезная умственная отсталость. Овладение коммуникативным языком невозможно (Beckung et al., 2004).