Эпилепсии с миоклоническими припадками у новорожденных, младенцев
Эпилепсии, характеризующиеся главным образом истинными миоклоническими припадками, т.е. припадками, проявляющимися клинически очень кратковременными резкими мышечными сокращениями по типу шоковых, а на ЭЭГ — быстрыми комплексами спайк-волна или полиспайк-волна, начинаются в младенчестве или раннем детстве (Aicardi, 1996; Guerrini, Aicardi, 2003; Arzimanoglou et al., 2004).
Такие эпилепсии часто путают с синдромом Леннокса-Гасто из-за часто повторяющихся коротких атак, которые могут быть причиной многократных падений, наличия разрядов типа спайк-волна при обоих состояниях и частой ассоциации с умственной отсталостью или ухудшением с припадками. Однако клинические и ЭЭГ-признаки миоклонических эпилепсий отличаются от проявлений СЛГ, и представление о контроле припадков и психическом развитии не всегда единообразны.
Миоклонические припадки представляют собой внезапные молниеносные мышечные сокращения; могут вовлекаться мышцы всего тела (массивная миоклоническая атака) или только верхних конечностей, лица или век. Судороги обычно симметричны, но могут быть односторонними или локализованными в небольших группах мышц. На иктальной ЭЭГ выявляются пароксизмы полиспайк-волн.
Иктальная электромиография демонстрирует очень короткие мышечные сокращения, за которыми следует период покоя в течение 100-350 мс. При относительной продолжительности этого периода расслабление мышц проявлется клинически, вызывая миоатонический припадок (Oguni et al., 1994, 1997). Иногда при так называемом «негативном миоклонусе» заметна только атоническая фаза (Guerrini и Aicardi, 2003).
Клинически, атония различной интенсивности и длительности часто сопровождается миоклонической судорогой. Генерализованная и достаточно длительная атония приводит к падениям (дроп-атакам) и по этой причине отсутствует возможность отличить по клиническим признаками атонический припадок от миоклонического. Поэтому миоклоническую-астатическую эпилепсию можно считать вариантом миоклонической эпилепсии, при котором особенно выражен атонический компонент.
Нозология миоклонических эпилепсий остается запутанной. Международная классификация выделяет три большие группы: тяжелые миоклонические эпилепсии или синдром Драве (Dravet et al., 1986, 1992а, 2005) (см. выше), доброкачественная миоклоническая эпилепсия (Dravet et al., 1992; Dravet, Bureau 2005), и эпилепсия с миоклоническими-астатическими припадками (Doose, 1992), к которым можно добавить неклассифицируемые случаи.
а) Доброкачественная миоклоническая эпилепсия младенцев. Доброкачественная миоклоническая эпилепсия (ДМЭ) характеризуется короткими миоклоническими припадками, спонтанными или вызванными шумом или контактом, начинающимися в возрасте от четырех месяцев до трех лет у детей с нормальным нервным развитием, преимуществен но у мальчиков (Dravet et al., 1992, 2005).
Миоклонические припадки—единственный тип припадков при этом заболевании, за исключением редких простых фебрильных судорог у некоторых пациентов. Интериктальная ЭЭГ, включая регистрируемую во время сна, нормальна, спонтанные разряды спайк-волна встречаются редко. Миоклониям на ЭЭГ сопутствуют разряды в форме быстрых генерализованных спайк-волн или полиспайк-волн с частотой более 3 Гц, начинающиеся и заканчивающиеся вместе с миоклониями.
Течение болезни доброкачественное, хорошо поддается монотерапии вальпроатом натрия, при необходимости в сочетании с этосуксимидом или бензодиазепинами. Лечение продолжается три или четыре года. Однако у отдельных детей сохраняются некоторые проблемы с учебой и легкая задержка развития. Фактически, далеко не всегда можно прогнозировать доброкачественное течение болезни, так как более чем у 10% больных сохраняются поведенческие и некоторые когнитивные нарушения (Dravet и Bureau, 2005).
Неясно, должен ли термин «доброкачественная миоклоническая эпилепсия» ограничиваться случаями с исключительно миоклоническими припадками. Aicardi и Levy Gomes (1989) сообщали о 19 детях, преимущественно мальчиках, с высокой частотой припадков в предшествующих поколениях, у которых наблюдались нечастые тонико-клонические припадки и/или короткие абсансы вместе с частыми миоклоническими судорогами и относительно благоприятным исходом болезни. Эти случаи подтверждают существование спектра не вызванных какими-либо поражениями, вероятно, генетически детерминированных миоклонических эпилепсий.
У некоторых младенцев припадки вызываются внезапными экстрацептивными или проприоцептивными стимулами. При данной «тактильной» или рефлекторной миоклонической эпилепсии (Ricci et al., 1995) прогноз благоприятный, и можно обойтись без лечения. Сходные с ДМЭ случаи могут возникать и у старших детей, и можно считать ДМЭ ранним проявлением идиопатической генерализованной эпилепсии (Arzimanoglou et al., 2004), эквивалентом ювенильной миоклонической эпилепсии, как предложили Dravet и Bureau (2005).
Генетические причины доброкачественной миоклонической эпилепсии (ДМЭ) неизвестны. Случаи заболевания редки, и не описано ни одного случая семейной заболеваемости. Arzimanoglou et al. (1996) описали семью, в которой у пробанда была эпилепсия с миоклоническими-астатическими припадками, а его младший брат перенес типичную ДМЭ с благоприятным исходом.
Атонический припадок.
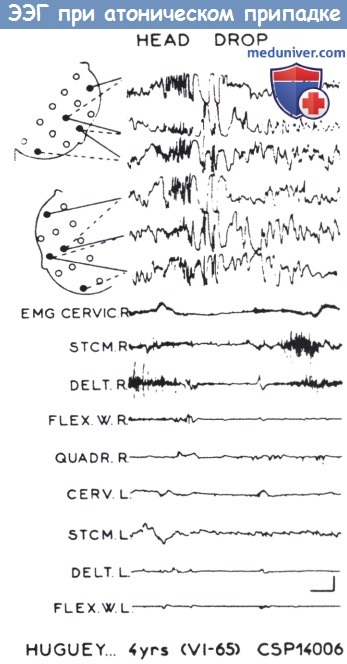
На ЭЭГ разряды начинаются вспышками быстрого ритма, за которыми следуют несколько комплексов спайк-волно и медленные волны.
На ЭМГ видно исчезновение нормальной тонической активности во время разрядов на ЭЭГ в задней группе мышц шеи (CERVIC.R), грудинноключично-сосцевидной мышце (STCM.R) и дельтовидной мышце (DELT.R).
HEAD DROP — свисание головы.
б) Миоклоническая-астатическая эпилепсия. Миоклоническую-астатическую эпилепсию (возможно, более точно эпилепсию с миоклоническими-астатическими припадками), вероятно, лучше отнести к категории генерализованных, не связанных со структурными поражениями миоклонических эпилепсий, а не к отдельным синдромам (Guerrini и Aicardi, 2003; Arzimanoglou et al., 2004; Guerrini et al., 2005). Термин «миоклоническая-астатическая» впервые использовали Doose et al. (1970) в публикации о «центроцефальном миоклоническом-астатическом petit mal» и позже развили Doose и Baier (1987) в статье «Генетические факторы эпилепсий с первивично генерализованными малыми припадками».
Первоначально так определяли форму наследственной, не связанной с повреждениями генерализованной эпилепии, и, возможно, включали случаи, которые сейчас называют тяжелыми или доброкачественными миоклоническими эпилепсиями, так же как и случаи, соответствующие современной концепции миоклонической-астатической эпилепсии (Guerrini и Aicardi, 2003).
Заболевание начинается позже, чем ДМЭ или синдром Драве, обычно в возрасте от одного года до пяти лет, чаще у мальчиков. Оно характеризуется клинически преобладанием чисто миоклонических или/и миоастатических припадков; припадки могут вызывать падения или, если припадок кратковременный, серию эпизодов свисания головы оседания на колени. Часто этим типам припадков сопутствуют другие, включая генерализованные тонико-клонические атаки, атипичные абсансы и эпизоды неконвульсивного эпилептического статуса.
Тонические припадки не являются преобладающими, но в некоторых сериях наблюдаются относительно часто (Kaminska et al., 1999; Oguni et al., 2001a). Позднее авторы отметили, что они возникали максимум в двух третях их случаев. Астатические припадки (дроп-атаки) могут иметь различные механизмы (астатические, миоклонические или тонические), но их невозможно определить без полиграфической записи. Тонические припадки могут более часто наблюдаться у детей с неблагоприятным исходом, но они также наблюдаются почти у 30% детей с благоприятным исходом заболевания (Kaminska et al., 1999).
На ЭЭГ выявляются быстрые (<3 Гц) генерализованные кратковременные (обычно меньше 4-5 секунд) разряды спайк-волна или полиспайк-волна. Doose подчеркивает наличие бипариетальных тета-ритмов.
Возможна ремиссия почти в 60% случаев в течение нескольких лет с нормальными когнитивными функциями (Oguni et al., 2001а; Guerrini et al., 2005). Однако в начале заболевания могут возникнуть трудности контроля падений. Они могут стать причиной травм и являются серьезной практической проблемой. У некоторых пациентов заболевание протекает тяжело, с частыми припадками и нарушениями обучения (20%) или умственной отсталостью (20%), но в начале заболевания исход предсказать невозможно.
Эффективность антиэпилептических препаратов вариабельна. Препаратом первого выбора считается вальпроат натрия или, если необходимо, комбинация вальпроата натрия и ламотриджина. При миоклони-ческих припадках и абсансах доказана эффективность этосуксимида и бензодиазепинов, особенно клобазама. Наконец, интересной альтернативной могут быть топи-рамин и леветирацетам. Следует избегать назначения карбамазепина, вигабатрина, окскарбазепина и габа-пен гина (Arzimanoglou et al., 2004; Guerrini et al., 2005).
в) Неклассифицированные формы. Ряд случаев миоклонических эпилепсий у младенцев и в раннем детском возрасте остается неклассифицированными, также существуют промежуточные между разными синдромами формы (Arzimanoglou et al., 2004). Их нужно отделять от других синдромов с частыми короткими атаками и повторяющимися падениями, особенно синдрома Леннокса-Гасто. Термин «миоклоническая-астатическая эпилепсия» все еще не имеет четких клинических или ЭЭГ признаков, он произвольно и с трудом дифференцируется от других миоклонических форм и может быть применен ко многим миоклоническим эпилепсиям. Дегенеративные миоклонические эпилепсии с прогрессивным течением обсуждаются в отдельных статьях на сайте.