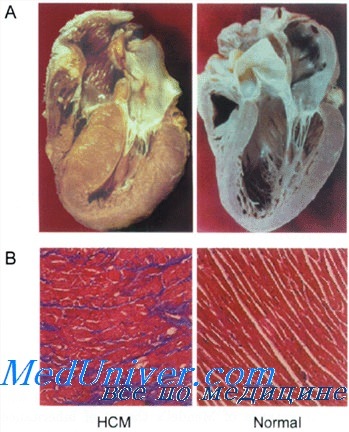
Предрасположенность к аритмиям при ГКМП. Титин при гипертрофической кардиомиопатии
Для изучения факторов, влияющих на подверженность к аритмиям при ГКМП, использовали мышиные модели. Было высказано предположение, что дезорганизация и фиброз миоцитов, которые считаются характерными патологическими особенностями ГКМП, являются факторами, повышающими подверженность желудочковым аритмиям и риску ВС.
Однако взаимосвязи между дезорганизацией фибрилл или фиброзом и склонностью к аритмиям на мышиной модели ГКМП обнаружить не удалось. В противоположность этому гипертрофия и повышенная сократимость коррелировали со склонностью к индуцированным аритмиям. Эти результаты позволяют предположить, что терапевтическое воздействие, ослабляющее рост миоцитов при ГКМП, также способно уменьшить риск сердечных аритмий.
Сердечный миозинсвязывающий протеин-С (МуВРС), белок с молекулярной массой 137 кДа, колируемый локализованным на хромосоме 11 геном MYBPC3, обеспечивает структурную целостность саркомера за счет связывания тяжелой МНС и титина (см. далее) и модулирует АТФазную активность миозина и сократимость сердечной мышцы в ответ на стимуляцию адренергических рецепторов. Совокупностью миссенс-мутаций, мутаций, затрагивающих участок сплайсинга, и вставок/делеций можно объяснить 40% случаев ГКМП.
Обычно при мутациях МуВРС клиническое течение ГКМП мягче, а выживаемость выше, чем при мутациях в тяжелой бета-цепи МНС. У значительной части пациентов, имеющих мутации МуВРС, клинические проявления ГАЖ не диагностируют до 50 лет и старше. С этим наблюдением согласуется наличие ассоциации между поздним проявлением признаков ГКМП и мутациями гена МуВРС.
У генетически модифицированных мышей, экспрессирующих укороченный вариант гена МуВРС, проявляются признаки ГКМП человека с поздним развитием гипертрофии миокарда, дезорганизацией миофибрилл и фиброзом. Мыши с такой мутацией невосприимчивы к аритмиям в отличие от животных, имеющих мутацию в тяжелой МНС, что подтверждает важность генотипа в детерминации подверженности развитию аритмий.
Если укороченный за счет мутации белок МуВРС заменить на белок МуВРС, который не способен фосфорилироваться в ответ на физиологические сигналы, у мышей все равно развивается ГКМП. Эти данные свидетельствуют о том, что для нормального функционирования миокарда необходимо фосфорилирование МуВРС.
Причиной гипертрофической кардиомиопатии служат мутации молекулы титина — гигантского белка с молекулярной массой 3 кДа, кодируемого локализованным на хромосоме 2 геном TTN. Располагаясь в саркомере между Z-и М-дисками, титин, вероятно, служит матрицей для сборки сократительных филаментов и обеспечивает их эластичность за счет серии элементов, подобных пружине.
Высокая стоимость анализа этого огромного гена служит препятствием для широкого внедрения популяционного скрининга ГКМП. Фенотипические проявления некоторых мутаций гена ТТN, ассоциированных с ГКМП, неотличимы от мутаций других генов саркомерных белков.
Регуляторная легкая цепь (кодируемая геном MYL2) и эссенциальиая легкая цепь (кодируемая геном MYL3) миозина принадлежат к белкам так называемого суперсемейства EF-руки (термин «EF-рука» широко используют при описании структуры Са2+-связывающих белков.), в молекуле которых находится мотив, имеющий вид «спираль-петля-спираль», который детерминирует скорость и силу скольжения акто-миозина путем взаимодействия с шарнирным устройством, соединяющим головку и стержень тяжелых MHС.
Мутации легких цепей миозина редко служат наследственной причиной ГКМП и обусловливают < 5% всех случаев заболеваний. Описана одна мутация эссенциальной легкой цепи миозина (Met149Val), сопряженная с отличительной морфологической особенностью ГКМП, а именно со срединно-желудочковой гипертрофией, ассоциированной с нарушениями гемодинамики. При мутации эссенциальной легкой цепи миозина меняется механочувствительный ответ (обусловленный ионными каналами, активируемыми растяжением),который является важнейшим свойством мышц; при этом возникают мощные колебательные движения и регионарные сокращения сердца, особенно важные для функционирования сосочковых мышц миокарда.