После идентификации в 1995-1996 гг. первых трех генов СУ QT (LQT1, LQT2, LQT3) были установлены и другие гены, связанные с СУ QT (LQT4-LQT10). В действительности не все эти гены могут быть ответственными за развитие СУ QT. Предполагают, что гены LQT4 и LQT7 (синдром Andersen-Tawil) вызывают комплексные клинические нарушения, при которых удлинение интервала QT — вторичный феномен, и его не следует расценивать как часть СУ QT.
Гены LQT5, LQT6, LQT8 хотя и редко, ответственны за возникновение СУ QT. Для генов LQT9 и LQT10 существуют только предварительные определения, и в настоящее время их следует расценивать как гипотетические генетические варианты СУ QT.
С эпидемиологической точки зрения гены LQT1, LQT2, LQT3 более чем на 90% ответственны за СУ QT, в то время как оставшиеся гены — за меньшее количество случаев. Другое интересное наблюдение заключается в том, что процент пациентов с СУ QT более чем с одной мутацией (часто определяемое как смешение гетерозигот) может достигать 4%.
Главное свойство всех идентифицированных в настоящее время генов СУ QT: кодирование субъединиц ионных каналов сердца (как в случае недавно описанного CAV3) есть нечто иное (изменение ионных токов реполяризации). Следовательно, термин «каналопатия» означает группу заболеваний, при которых имеет место СУ QT.
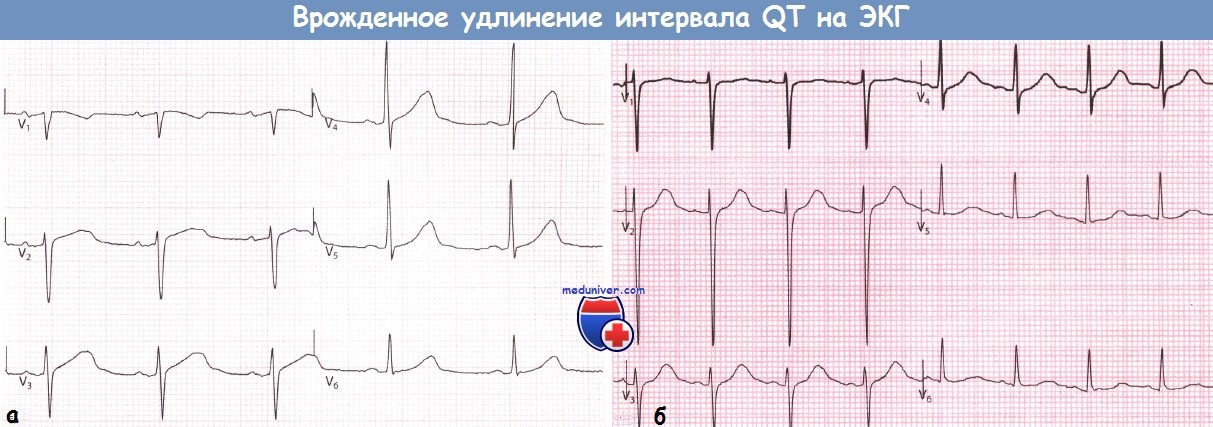
Врожденное удлинение интервала QT.
ЭКГ зарегистрированы у не имеющего симптомов отца (а) и его дочери, испытывающей серьезные симптомы (б).
Лечение синдрома удлиненного интервала QT
Рекомендации по ведению пациентов с СУ QT были недавно обобщены в объединенных Рекомендациях АСС/AHA/ESC по желудочковым аритмиям.
С середины 1970-х гг. связь между развитием сердечных событий и гиперсимпатикотонией создала основу для применения бета-адреноблокаторов при лечении болезней. Эффективность бета-адреноблокаторов у пациентов с СУ QT была вновь доказана в масштабных исследованиях, которые продемонстрировали, что общая смертность составляет < 2% при наблюдении > 5 лет. Бета-AБ особенно эффективны у пациентов при наличии гена LQT1.
В этой группе комбинированный показатель частоты распространения успешной реанимации при остановке сердца и внезапной сердечной смерти был равен только 1%. Этот показатель увеличивался до 7% у пациентов с геном LQT2 и до 14% — у пациентов с LQT3. Поскольку риск остановки сердца и ВСС у бессимптомных пациентов в возрасте < 40 лет, с четко удлиненным корригированным интервалом QTc и не получавших лечения достигает 13%, то всех их следует лечить.
Небольшое исключение составляют в основном мужчины в возрасте 25-30 лет, с пограничными значениями QTc и геном LQT1. Всех пациентов с наличием симптомов также необходимо лечить. Для 20-30% пациентов, у которых могут быть повторные обмороки, несмотря на применение бета-ЛП, наиболее приемлемым метолом лечения может стать левосторонняя симпатическая денервация сердца или профилактическая установка имплантируемого кардиовертера-дефибриллятора.
Анализ сердечных событий в зависимости от генотипа показал, что у пациентов с генами LQT2 и LQT3 с продолжительностью интервала QTc > 500 мсек и возникновением первого эпизода синкопе в возрасте < 7 лет является независимым предиктором неэффективности бета-АБ.
У этих пациентов следует рассмотреть возможность профилактического применения ИКД. Имплантацию постоянного электрокардиостимулятора сегодня используют редко: только у пациентов с атриовентрикулярной (АВ) блокадой или с брадикардией либо с паузозависимыми тахиаритмиями как дополнение к бета-АБ и после рассмотрения возможности установки ИКД.
Известно, что ген LQT3 связан с увеличением позднего натриевого тока, и в 1995 г. это положение было проверено фармакологически. Действительно, блокада канала натрия мексилетином сокращала интервал QT у пациентов с геном LQT3, но не с LQT2. Растущее число отдельных сообщений свидетельствует о возможной эффективности мексилетина в комбинации с бета-АБ у младенцев с геном LQT3, со значительным удлинением интервала QT и выраженными аритмиями.
Поскольку в течение нескольких лет генотипирование применяли в основном в качестве научных исследований, выявление сегодня скрытых мутаций и их роли в генезе аритмий должно существенно повлиять на клиническое ведение больных. Учитывая высокую вероятность (70%) идентификации вызывающей болезнь мутации у пробанда, необходимо принять меры к быстрому проведению скрининга членов семьи и профилактическим мероприятиям. Таким образом, молекулярный скрининг должен стать частью обычного клинического ведения пациентов с СУ QT. Генотипирование эффективно с точки зрения затрат; стратегии доступны и по времени, и по стоимости молекулярного скрининга.