Расшифровка генома - кратко с точки зрения внутренних болезней
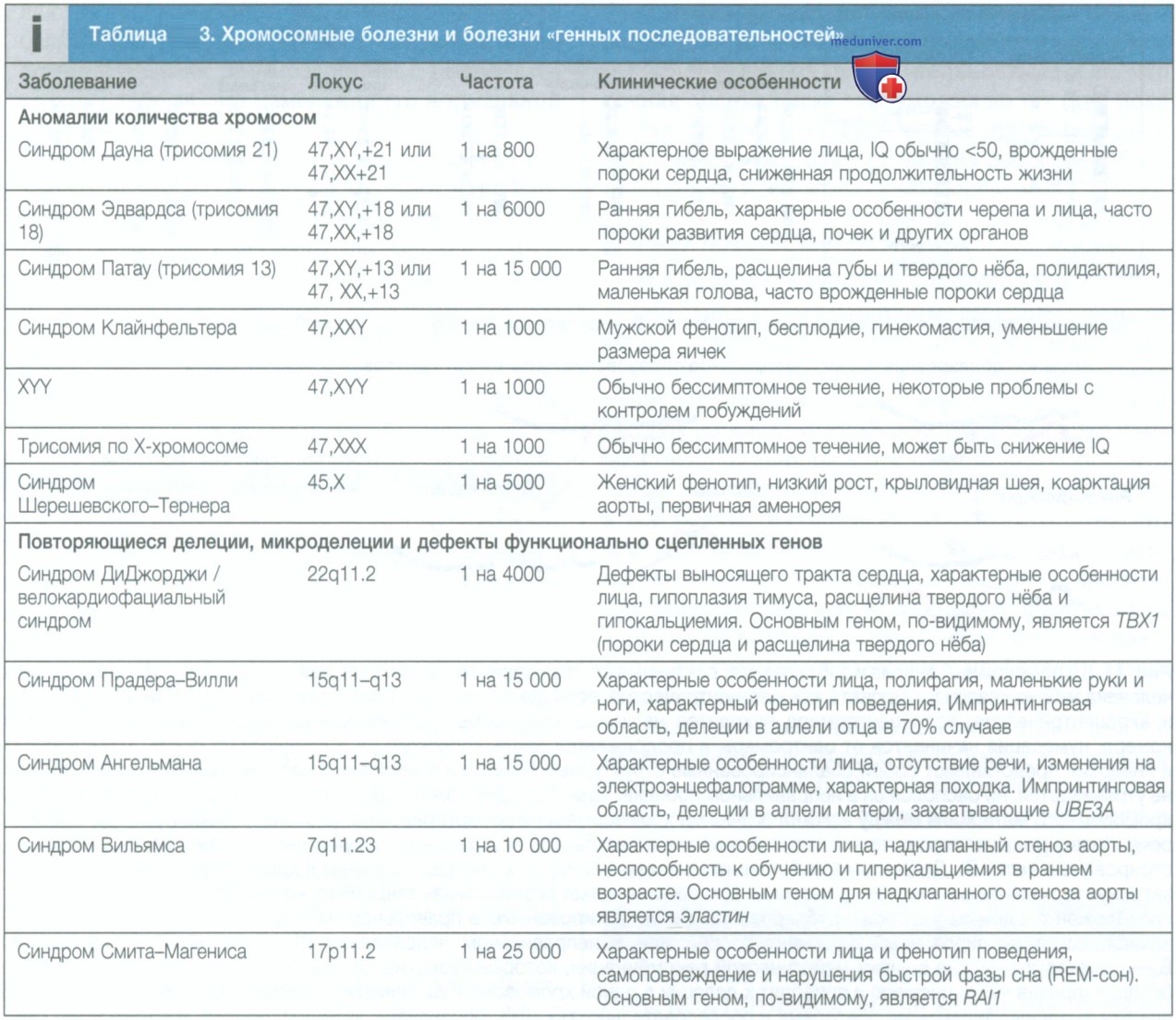
а) Изучение хромосом. На протяжении десятилетий основой клинического цитогенетического анализа был анализ хромосом в метафазе с использованием световой микроскопии. Целью этого анализа было обнаружить появление или утрату целых хромосом (анеуплоидия) или крупных хромосомных сегментов (>4 млн пар оснований). Недавно хромосомный анализ уступил место полногеномному микроматричному анализу [матриксная сравнительная геномная гибридизация (comparative genomic hybridization)], который позволяет быстрее и точнее обнаружить появление новых сегментов ДНК или утрату имеющихся во всем геноме (см. табл. 3).
Микроматрицы представляют собой решетки с многочисленными лунками, которые содержат короткие последовательности ДНК (эталонная ДНК), комплементарные известным последовательностям генома.
Эталонную ДНК и ДНК пациента маркируют цветными флюоресцентными красителями (ДНК пациента обычно маркируют зеленым флюоресцентным красителем, а эталонную ДНК — красным) и вносят в решетку микроматрицы. При наличии одинакового количества связанных с пятном ДНК пациента и эталонной ДНК отмечается желтая флюоресценция. При избытке ДНК пациента (представляющей собой дублированные участки хромосомы) пятно будет более зеленым. В случае когда соотношение эталонной ДНК и ДНК пациента составляет 2 к 1, пятно будет более красным (выглядит как оранжевое), что соответствует гетерозиготной делеции участка хромосомы (рис. 1).
Рисунок 1. Выявление хромосомных нарушений при помощи сравнительной геномной гибридизации. Делеции и дупликации обнаруживаются как отклонение соотношения количества ДНК пациента и эталонной ДНК в микроматрице от 1:1. Соотношение, превышающее 1, свидетельствует о дупликации, тогда как соотношение ниже 1 указывает на наличие делеции
Матриксная сравнительная геномная гибридизация и другие методы, основанные на использовании матрицы, позволяют обнаруживать небольшие хромосомные делеции и дупликации. Кроме того, эти методы, как правило, имеют большую чувствительность в обнаружении мозаицизма, чем обычное кариотипирование (при наличии мозаицизма существуют две или более популяции клеток с разными генотипами, которые были получены из одной оплодотворенной яйцеклетки).
В то же время методы, основанные на использовании матрицы, не позволяют обнаруживать сбалансированную хромосомную перестройку, при которой отсутствует утрата или увеличение генного/хромосомного материала, как происходит, например, при сбалансированных реципрокных транслокациях или при увеличении общего числа копий, в частности при триплоидии.
Широкое применение методов, основанных на использовании матрицы, повлекло за собой ряд проблем в клинической интерпретации, включая идентификацию вариаций числа копий (сору number variant) с неопределенной клинической значимостью, вариации числа копий с вариабельной пенетрантностью и случайные результаты. Вариация числа копий с неопределенной клинической значимостью представляет собой вариант потери или прибавления хромосомного материала, для которого нет достаточных данных, чтобы сделать вывод о связи мутации с неспособностью к обучению и/или с медицинскими проблемами.
Такая неопределенность представляет собой трудности при обследовании семей и может спровоцировать выраженную тревожность. Тем не менее в дальнейшем по мере формирования более крупных баз данных о вариациях числа копий, вероятно, появится больше ясности.
Вариации числа копий с вариабельной пенетрантностью, также известные как локусы нейрочувствительности, характеризуются делецией или дупликацией хромосом, при которых снижается порог для развития неспособности к обучению или расстройств аутистического спектра. Вариации числа копий с вариабельной пенетрантностью чаще обнаруживаются у лиц с неспособностью к обучению и/или расстройствами аутистического спектра, чем в общей популяции. В настоящее время считают, что на фенотипическую экспрессию этих локусов нейрочувствительности должны влиять дополнительные модифицирующие факторы (генетические факторы, факторы окружающей среды или стохастические факторы).
Наконец, случайные результаты представляют собой делецию или дупликацию в гене или генах, которые связаны с фенотипом или риском, не имеющими отношения к жалобам пациента. Например, если при матриксной сравнительной геномной гибридизации по поводу умственной отсталости обнаруживается делеция в гене BRCA1, это будет считаться случайным результатом.
б) Изучение генов:
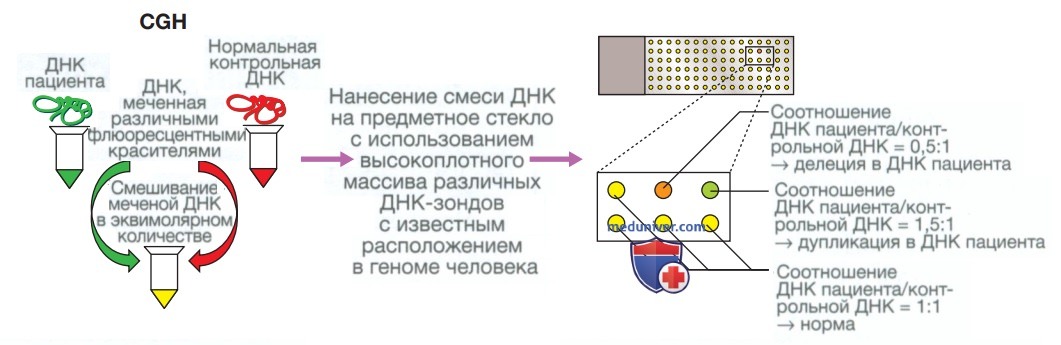
1. Амплификация генов: полимеразная цепная реакция. Полимеразная цепная реакция представляет собой фундаментальный метод лабораторных исследований, в ходе которого выполняется амплификация целевых участков генома человека, которые используются для дальнейшего анализа, чаще всего для секвенирования ДНК. При полимеразной цепной реакции выполняется циклическое изменение температуры: повторные циклы нагревания и охлаждения обеспечивают исходное разделение двухцепочечной ДНК на две отдельные цепи (процесс, называемый денатурацией). Каждая из полученных цепей служит матрицей на последующем этапе репликации, во время которого для отжига в конкретной области генома используются праймеры.
Такие циклы нагревания/охлаждения и денатурации/репликации многократно повторяются, в результате чего происходит экспоненциальная амплификация ДНК между сайтами праймера (рис. 2).
Рисунок 2. Полимеразная цепная реакция. Небольшое количество ДНК пациента добавляют в реакционную смесь, содержащую праймеры (короткие олигонуклеотиды длиной 18-21 пары оснований, связывающиеся с ДНК, прилежащей к представляющей интерес области), дезоксинуклеотидфосфаты (дАТФ, дЦТФ, дГТФ, дТТФ), которые используются для синтеза новой ДНК, и термостабильную ДНК-полимеразу. Сначала реакционную смесь нагревают до 95 °C, при этом происходит расхождение двухцепочечной молекулы ДНК. Затем реакционную смесь охлаждают до 50-60 °C, что позволяет праймерам связываться с ДНК-мишенью. После этого реакционную смесь нагревают до 72 °C, в результате чего ДНК-полимераза начинает образовывать новые цепи ДНК. Эти циклы повторяются 20-30 раз, что приводит к экспоненциальной амплификации фрагмента ДНК между сайтами праймера. Полученные продукты полимеразной цепной реакции могут быть использованы для дальнейшего анализа, чаще всего для секвенирования ДНК (см. рис. 3)
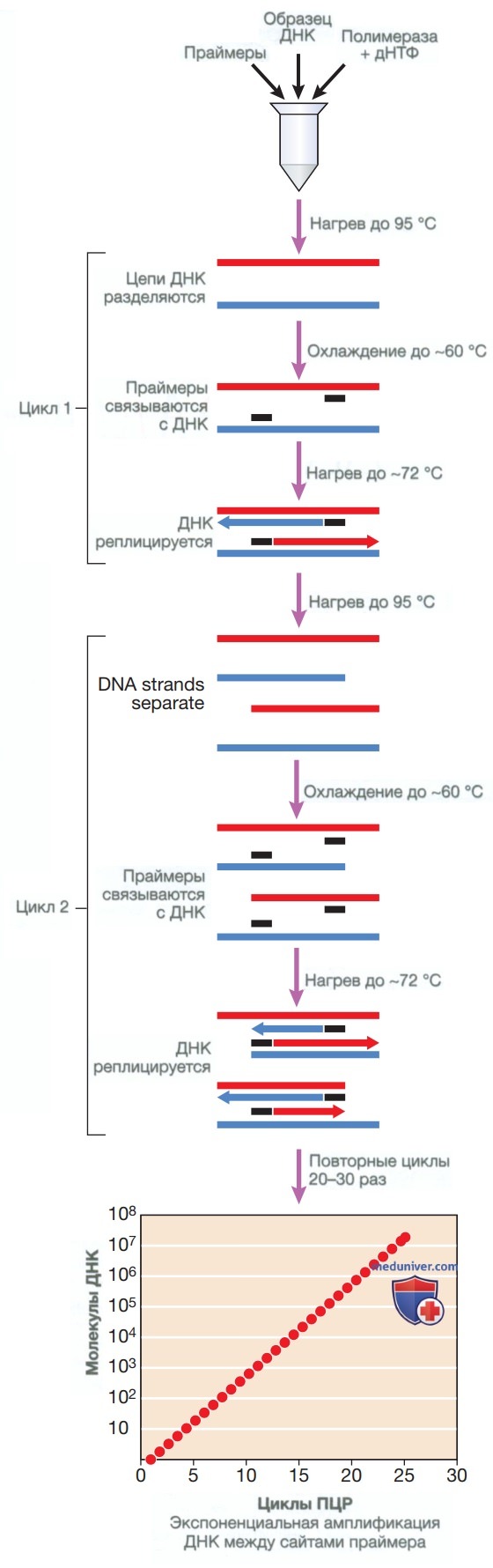
2. Секвенирование генов. В середине 1970-х годов ученый Фредерик Сенгер впервые использовал технологию секвенирования ДНК (секвенирование по методу Сенгера), при помощи которой были определены точный порядок и тип нуклеотидов (тимин, цитозин, аденин и гуанин) в молекуле ДНК. В современном секвенировании по методу Сенгера используются терминирующие нуклеотиды с флюоресцентной меткой, которые последовательно включаются во вновь синтезированную цепь ДНК, в результате чего образуется множество цепей ДНК различной длины. В дальнейшем выполняется капиллярный электрофорез этих цепочек ДНК, в ходе которого они сортируются по размеру.
Это позволяет провести считывание фрагментов с использованием лазера и получить хроматограмму последовательности, которая соответствует последовательности мишени (рис. 3). Хотя секвенирование по методу Сенгера было революционным, его масштабирование оказалось затруднительным и дорогостоящим. Об этом свидетельствует проект «Геном человека», в котором на секвенирование всего генома человека ушло 12 лет, а затраты приблизились к 3 млрд долларов.
Рисунок 3. Секвенирование по методу Сенгера, очень широко использующееся в ДНК-диагностике. В данном методе применяются фрагменты ДНК, амплифицированные при помощи полимеразной цепной реакции и соответствующие гену, который представляет интерес. Реакцию секвенирования проводят с помощью комбинаций дНТФ и ди-дезокси-дНТФ (ддАТФ, ддТТФ, ддЦТФ и ддГТФ) с флюоресцентной меткой, которые включаются во вновь синтезированную ДНК, что приводит к терминации цепи в этой точке. Затем продукты реакции подвергаются капиллярному электрофорезу, и фрагменты различного размера обнаруживают при помощи лазера. В результате этого получают хроматограмму последовательности, которая соответствует целевой последовательности ДНК
С недавним появлением группы технологий, известных под общим названием «секвенирование нового поколения» (next-generation sequencing — NGS; рис. 4), в секвенировании ДНК вновь произошел переворот. NGS относится к семейству технологий секвенирования, в которых используются те же пять основных принципов, что и в методе Сенгера.
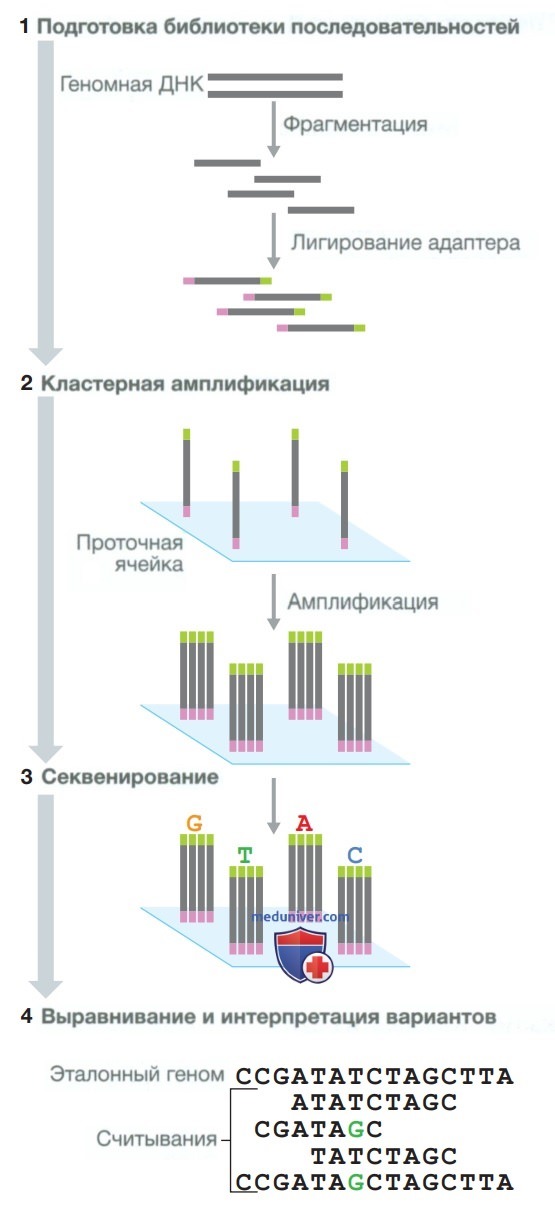
Рисунок 4. Секвенирование путем синтеза, используемое в системе компании Illumina. (1) Подготовка библиотеки последовательностей: ДНК фрагментируется, и специальные адаптеры связываются с фрагментированными концами. (2) Кластерная амплификация: библиотека загружается в проточную ячейку, и адаптеры гибридизируются с поверхностью проточной ячейки. Гибридизируется каждый связанный фрагмент. (3) Секвенирование. (4) Выравнивание и интерпретация вариантов: считанные последовательности выравниваются относительно эталонной последовательности с использованием сложного программного обеспечения; определяются различия между эталонным и исследуемым геномом
• Подготовка библиотеки последовательностей. Образцы ДНК фрагментируются (расщепляются под действием ферментов или ультразвукового воздействия), а затем модифицируются заданными адаптерными последовательностями.
• Амплификация. Фрагмент последовательности из библиотеки амплифицируется с образованием ДНК-кластеров, каждый из которых происходит от одного и того же фрагмента ДНК. Каждый кластер будет действовать как одна секвенирующая реакция.
• Захват. Если секвенируется весь геном, данный этап не проводится. Этап захвата требуется в случаях, когда необходимо целевое ресеквенирование, например при исследовании панели генов или изучении экзома (табл. 9).
• Секвенирование. Каждый ДНК-кластер секвенируется одновременно, и для каждого из них проводится сбор данных. Этот процесс известен как «считывание». Обычно секвенируется участок длиной от 50 до 300 последовательностей (подробное описание трех наиболее часто используемых методов секвенирования — синтеза, лигирования и ионного полупроводникового секвенирования — представлено в табл. 10).
• Выравнивание и идентификация вариантов. При помощи специального программного обеспечения проводятся анализ считанных последовательностей и сравнение полученных данных с эталонным шаблоном. Этот процесс известен как «выравнивание», или «картирование». Несмотря на то что геном человека содержит 3 млрд оснований, выравнивание позволяет необычайно точно определить геномное происхождение, если считываемый участок состоит из 25 или более нуклеотидов. Варианты выявляются при наличии различий между считанным и эталонным геномом.
Например, если в половине считанных последовательностей в определенном положении обнаруживается нуклеотид, отличный от такового в эталонном геноме, вероятно, имеется гетерозиготная замена основания. Количество считываний, выровненных в заданной точке, называется глубиной или охватом. Чем больше глубина считывания, тем точнее определяются варианты. Тем не менее в целом для получения диагностически значимых результатов достаточной считается глубина 30 и более считываний.
Вместо того чтобы единовременно секвенировать только одну небольшую последовательность ДНК, NGS позволяет анализировать сотни тысяч цепочек ДНК в одном эксперименте и поэтому часто также называется технологией множественного параллельного секвенирования. Современное оборудование для NGS позволяет секвенировать весь геном человека за 1 день при стоимости, приблизительно равной 1000 долларов США.
3. Захват при секвенировании нового поколения. Несмотря на то что теперь имеется возможность проводить секвенирование всего генома в одном эксперименте, оно не всегда является оптимальным использованием NGS. Захват при NGS относится к соосаждению целевой области генома и может затрагивать от нескольких генов до нескольких сотен генов, связанных с заданным фенотипом (панель генов), экзоны всех известных кодирующих генов (экзом) или экзоны всех кодирующих генов, для которых известна связь с заболеванием (клинический экзом).
Каждый из этих подходов направленного повторного секвенирования имеет ряд преимуществ и недостатков (см. табл. 9). Для применения NGS с оптимальной пользой для пациента важно, чтобы клиницист хорошо понимал, какой тест лучше использовать в конкретной клинической ситуации.
4. Проблемы, связанные с применением технологий секвенирования нового поколения. Геномные технологии способны произвести переворот в практической медицине, и более быстрое и более дешевое секвенирование ДНК увеличивает возможности диагностики, лечения и профилактики заболеваний. Тем не менее геномные технологии не лишены недостатков, одним из которых является хранение огромного количества данных, полученных при NGS. Хотя А, С, Т и G нашего геномного кода могут храниться в памяти смартфона, для хранения информации, необходимой для обработки генома каждого человека, нужны огромные компьютеры, способные хранить несколько петабайт данных (1 петабайт равен 1 млн гигабайт данных).
Даже если нам удастся успешно хранить и обрабатывать такие крупные базы данных, у нас должна быть возможность сортировать миллионы нормальных вариантов для выявления одной (или в редких случаях нескольких) патогенной мутации, которая вызывает заболевание. В определенной степени этого можно добиться путем применения сложных алгоритмов, однако для их разработки требуются время и значительный опыт, и в них могут быть ошибки.
Более того, даже после тщательного анализа этих данных специалистами по биоинформатике, вероятнее всего, у клиницистов останутся некоторые варианты, которые невозможно будет окончательно классифицировать как патогенные или непатогенные из-за отсутствия достаточного количества данных. Это может быть связано с тем, что у нас просто нет достаточных знаний о гене, поскольку конкретный вариант не был описан ранее и/или был выявлен у родителя, не имеющего заболевания. Такие варианты следует интерпретировать с осторожностью, и, как правило, в этих случаях требуется участие эксперта по генетике, поскольку интерпретацию нужно выполнять в контексте клинической картины, когда часто применяется подход «Невиновен до тех пор, пока не доказано обратное».
Наконец, если мы хотим провести анализ всего генома или даже экзома, можно предполагать, что мы регулярно будем получать «случайные» или вторичные результаты, т.е. результаты, не связанные с исходным диагностическим запросом. До настоящего времени в Великобритании применялся консервативный подход при оценке случайных результатов.
5. Применение секвенирования нового поколения. В настоящее время NGS часто используется в диагностических лабораториях для выявления замен оснований и инсерционно-делеционных мутаций (хотя обнаружение последних исходно проблематично). На сегодняшний день проблема NGS заключается в выявлении крупных делеций или дупликаций, затрагивающих несколько сотен или тысяч оснований и, следовательно, превышающих одно считывание. Тем не менее все чаще этот количественный анализ проводится с использованием сложных вычислительных методов, что избавляет от потребности в более традиционных технологиях, таких как матриксная сравнительная геномная гибридизация.
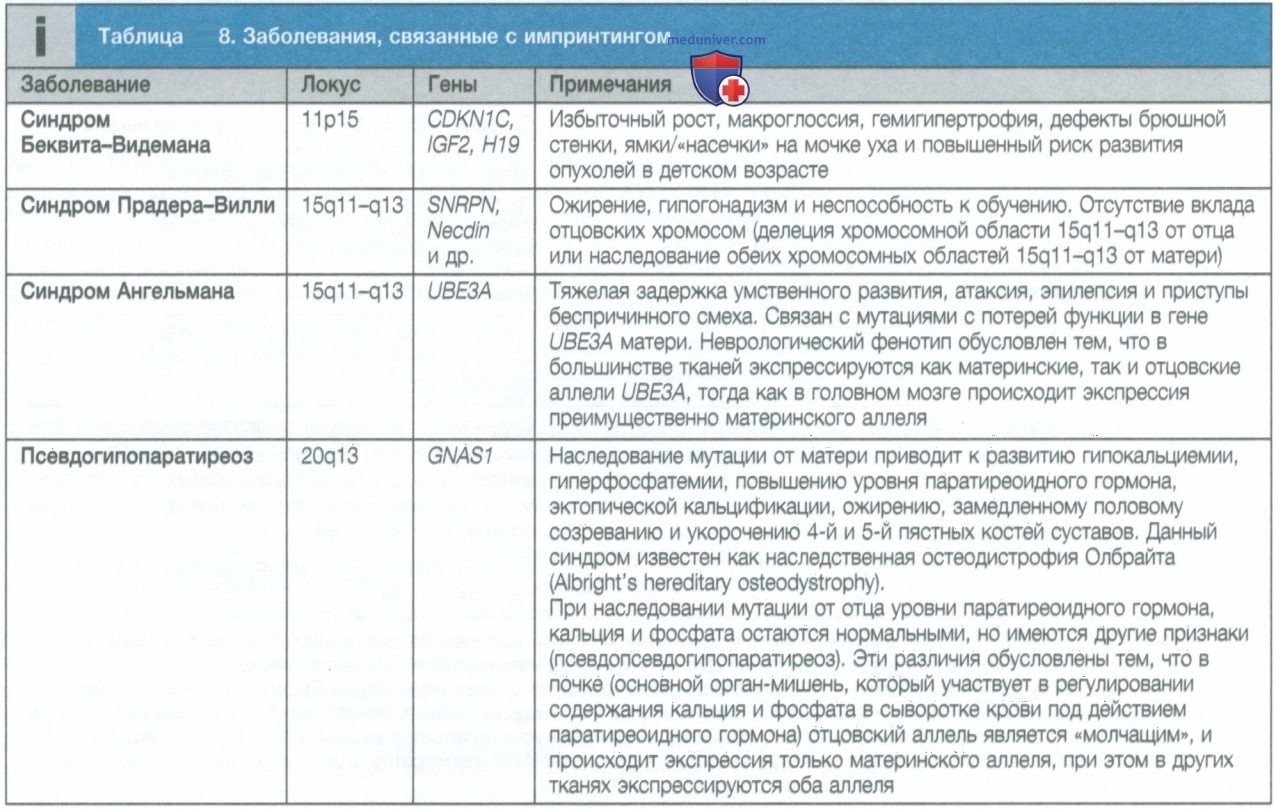
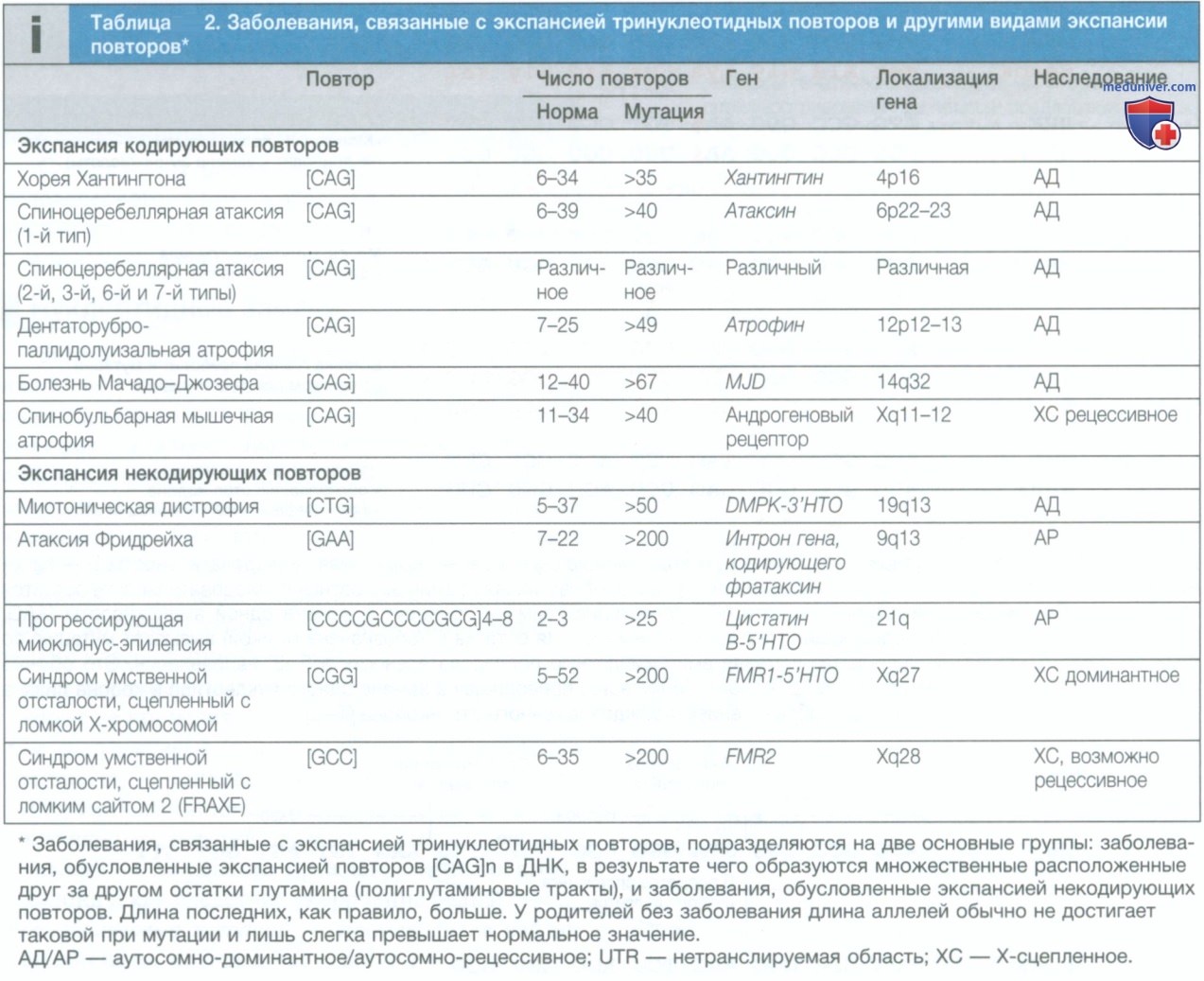
Дополнительным преимуществом применения NGS может быть обнаружение сбалансированных и несбалансированных транслокаций и мозаицизма, поскольку NGS оказалось чрезвычайно чувствительным при выявлении последних в случае высокого охвата считывания для данного региона. В то же время следует отметить, что с помощью NGS по-прежнему невозможно проводить исследование эпигенома (и поэтому выявлять состояния, обусловленные нарушением импринтинга, такие как синдромы Беквита—Видемана, Рассела—Сильвера, Ангельмана и Прадера—Вилли). Кроме того, NGS не позволяет диагностировать экспансии тринуклеотидных повторов, в частности болезнь Хантингтона, миотоническую дистрофию и синдром ломкой Х-хромосомы (см. табл. 8 и 2).
6. Секвенирование III поколения. Секвенирование III поколения или одномолекулярное секвенирование все чаще используется в диагностике. Как и в случае секвенирования нового или II поколения, в продаже имеется несколько различных платформ. Одной из наиболее успешных является технология SMRT (single-molecule sequencing in real time — одномолекулярное секвенирование в реальном времени), разработанная компанией Pacific Biosciences. В этой системе используется одноцепочечная молекула ДНК (в отличие от NGS, при котором применяются амплифицированные кластеры), которая играет роль матрицы для последовательного включения нуклеотидов с флюоресцентной меткой при участии полимеразы.
При добавлении каждого комплементарного нуклеотида до его удаления и добавления другого нуклеотида регистрируется флюоресценция (и, следовательно, идентичность нуклеотида).
Основным преимуществом секвенирования III поколения является большая длина полученных считанных последовательностей, которая достигает приблизительно 10—15 килобаз. Кроме того, данный метод дешевле, чем NGS, поскольку он требует меньшего количества реагентов. Учитывая эти преимущества, секвенирование III поколения, вероятно, в ближайшем будущем заменит NGS. Ввиду путаницы в терминах NGS и секвенирования III поколения эти технологии все чаще обозначают как «массовое параллельное секвенирование».
Видео методика и принципы ПЦР (полимеразной цепной реакции) в диагностике