Генетические заболевания - кратко с точки зрения внутренних болезней
Семейные генетические заболевания обусловлены конституциональными мутациями, которые наследуются через зародышевую линию клеток. Однако разные мутации в одном и том же гене могут иметь различные последствия, что зависит от генетического механизма, лежащего в основе заболевания. Около 1% людей в популяции имеют конституционные мутации, которые приводят к развитию заболевания.
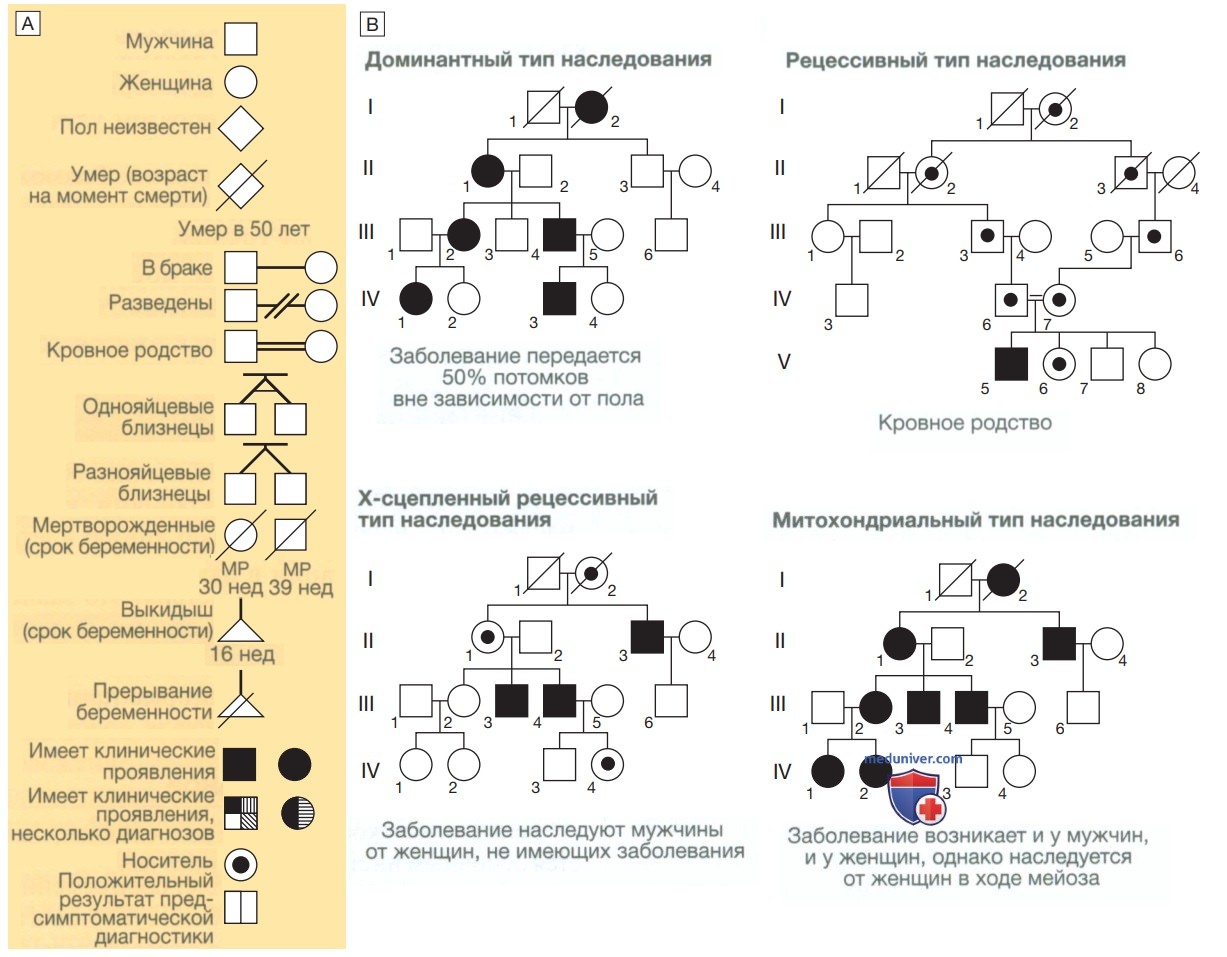
а) Составление родословной. Генеалогическое древо, или родословная, является основным инструментом клинического генетика, который рутинно осуществляет сбор семейного анамнеза в трех поколениях у обоих супругов и выясняет подробные сведения обо всех заболеваниях у членов семьи, кровном родстве, датах рождения и смерти и всех случаях невынашивания беременности или младенческой смерти. Основные символы и номенклатура, используемые при составлении родословной, показаны на рис. 1.
Рисунок 1. Составление родословной и характер наследования. А — основные символы, используемые для составления родословной в форме диаграммы. В — основные виды наследования заболеваний (подробности в тексте)
б) Характер наследования заболеваний:
1. Аутосомно-доминантное наследование. Потратьте некоторое время на составление следующей родословной.
Анна направлена к клиническому генетику с целью обсуждения истории заболевания раком толстой кишки, который был диагностирован у нее в возрасте 46 лет, и семейного анамнеза рака толстой кишки/рака эндометрия. У матери пациентки в возрасте 60 лет был выявлен рак эндометрия, а у двоюродной сестры после пятидесяти лет был обнаружен рак толстой кишки, при этом ее мать (тетя пациентки) здорова. Бабушка и дедушка пациентки по материнской линии умерли «от старости». Данных семейного анамнеза по отцовской линии нет. У отца есть один брат, и оба его родителя умерли «от старости» после восьмидесяти лет. У Анны есть две здоровые дочери в возрасте 12 и 14 лет и здоровая родная сестра.
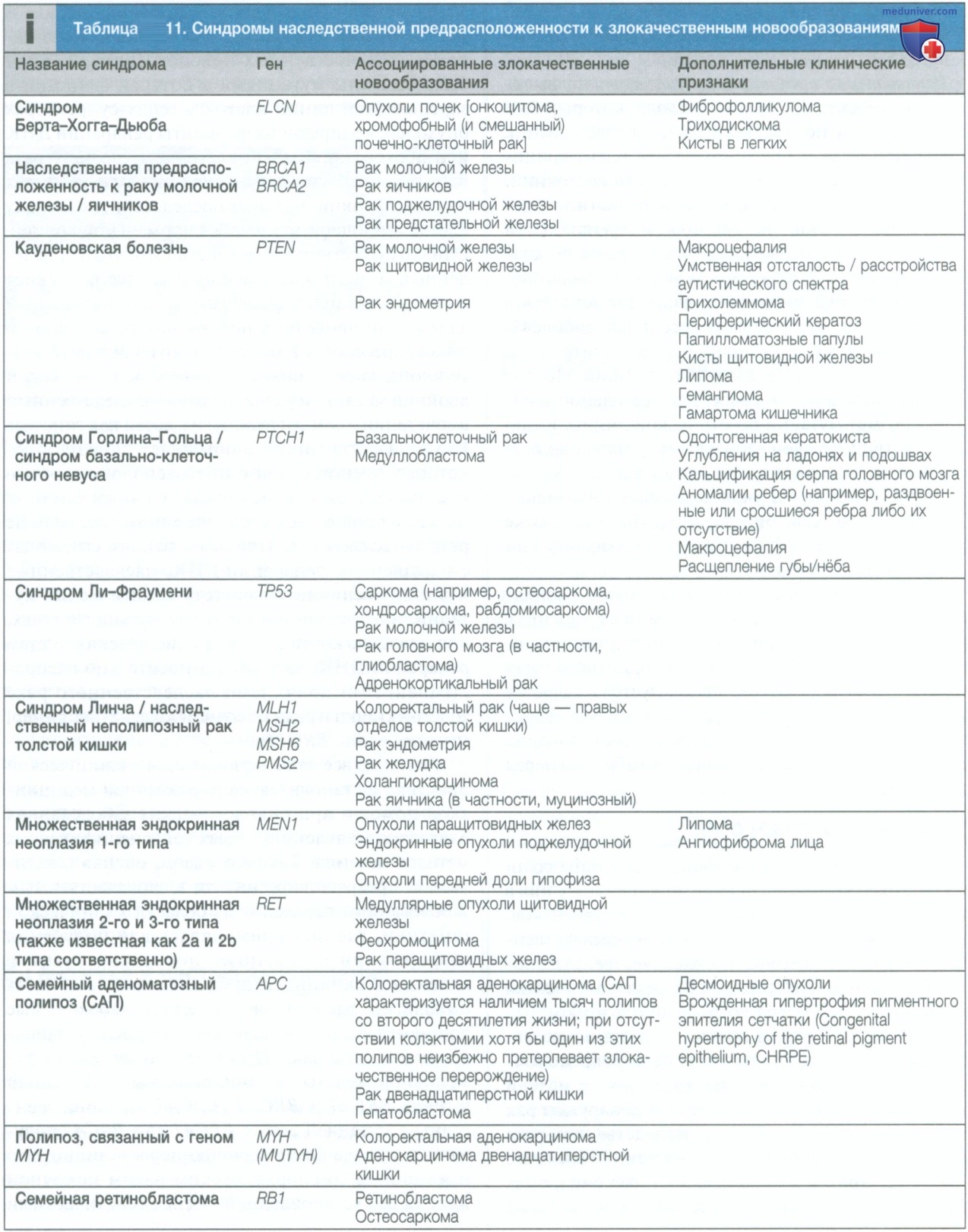
Такой семейный анамнез типичен для аутосомно-доминантного наследования (см. рис. 1): наблюдаемый в данном случае наследственный неполипозный рак толстой кишки, также известный как синдром Линча, связан с нарушением в одном из генов, ответственных за репарацию ошибочно спаренных оснований: MSH2, MSH6, MLH1 и PMS2 (см. табл. 11).
Аутосомно-доминантный тип наследования характеризуется следующими особенностями.
• В каждом поколении есть лица, у которых есть заболевание (за исключением мутаций, впервые возникших у человека с данным заболеванием).
• У мужчин и женщин заболевание, как правило, возникает примерно с одинаковой частотой (за исключением случаев, когда клиническая картина заболевания зависит от пола, например при наследственной предрасположенности к раку молочной железы и/или яичников).
Риск того, что дети унаследуют от родителя аутосомно-доминантное заболевание, составляет 1:2 (или 50%). Этот риск для потомства сохраняется для каждой беременности, поскольку половина гамет родителя (сперматозоиды или яйцеклетки) будет содержать поврежденную хромосому/ген, а другая половина — нормальную хромосому/ген.
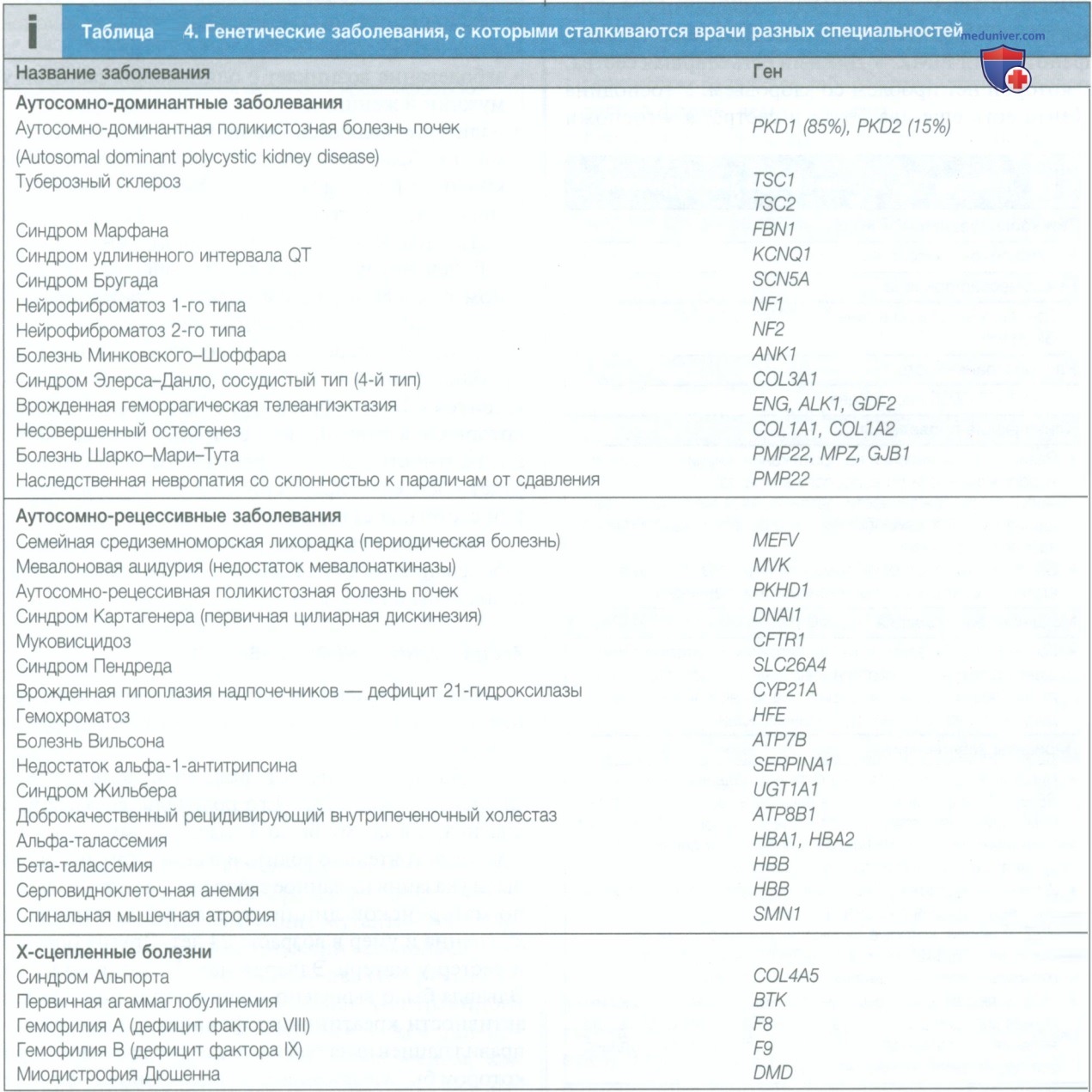
Существует большой список заболеваний, которые наследуются по аутосомно-доминантному типу. Некоторые из них представлены в табл. 4.
2. Аутосомно-рецессивное наследование. Составьте еще одно генеалогическое древо, в котором должно быть отражено следующее.
Супруги Кент, генетически не родственные, были направлены на генетическое обследование, поскольку у их сына Джейми возникло тяжелое заболевание печени в период новорожденности. Одним из исследований, проведенных детским гепатологом, было обследование на дефицит альфа-1-антитрипсина (табл. 5). Было обнаружено, что у Джейми фенотип PiZZ. Обследование подтвердило, что оба родителя являются носителями гена с фенотипами PiMZ. У Джейми есть старшая сестра, у которой нет проблем со здоровьем. У господина Кента есть еще два брата и сестра, а у госпожи Кент — младший брат. Все бабушки и дедушки живы и здоровы. В семейном анамнезе указаний на дефицит альфа-1 -антитрипсина нет.
Такой семейный анамнез характерен для аутосомно-рецессивного типа наследования (см. рис. 1), при котором для развития заболевания необходимо, чтобы оба аллеля гена были мутантными. Больной человек наследует по одному мутантному аллелю от каждого из своих родителей, которые, следовательно, являются здоровыми носителями этого гена. Заболевание с аутосомнорецессивным типом наследования можно заподозрить в семье, если:
• заболевание возникает с одинаковой частотой у мужчин и женщин;
• родители являются кровными родственниками, т.е. брак кровосмесительный. При наличии кровного родства мутации обычно являются гомозиготными, то есть один и тот же мутантный аллель наследуется от обоих родителей;
• заболевание развивается у братьев и сестер в одном поколении, таким образом, состояние может возникнуть совершенно неожиданно.
Аутосомно-рецессивное заболевание возникает приблизительно у 1 из 4 детей, рожденных от носителей гена. Таким образом, риск для детей, родители которых являются носителем гена, составляет 25%, а вероятность того, что ребенок без заболевания будет носителем при наличии заболевания у брата или сестры, составляет 2/3.
Примеры некоторых аутосомно-рецессивных заболеваний, обсуждаемых в других главах книги, приведены в табл. 4.
3. Х-сцепленное наследование. Ниже приведен пример родословной, характерной для Х-сцепленного рецессивного наследования (см. рис. 1).
У Эдварда диагностирована миодистрофия Дюшенна (табл. 6). Его родители заподозрили диагноз, когда ему было 3 года, потому что он не мог самостоятельно ходить и в семейном анамнезе были указания на данное заболевание: дядя Эдварда по материнской линии страдал миодистрофией Дюшенна и умер в возрасте 24 лет. Других братьев и сестер у матери Эдварда нет. После того как у Эдварда было выявлено значительное повышение активности креатининфосфокиназы, педиатр направил пациента на генетическое обследование, при котором была выявлена делеция экзонов 2-8 в гене дистрофина.
У Эдварда есть здоровая младшая сестра, бабушки и дедушки по линии отца и матери также здоровы, хотя у бабушки по материнской линии недавно появились признаки кардиомиопатии. Кроме того, у отца Эдварда есть здоровые старшие сестра и брат.
Генетические заболевания, вызванные мутациями в Х-хромосоме, имеют специфические особенности.
• Заболевания с Х-сцепленным типом наследования являются в основном рецессивными и развиваются у мужчин, имеющих мутантный аллель. Это связано с тем, что у мужчин есть только одна Х-хромосома, а у женщин — две (см. рис. 1). В то же время иногда признаки Х-сцепленного заболевания могут проявляться у женщин-носителей, что связано с таким феноменом, как искаженная инактивация Х-хромосомы. Все эмбрионы женского пола размером около 100 клеток стабильно инактивируют одну из двух своих Х-хромосом в каждой клетке.
Если эта инактивация носит случайный характер, примерно в половине клеток будут экспрессироваться гены из одной Х-хромосомы, а в другой половине клеток — из другой Х-хромосомы. При наличии мутантного гена часто происходит «уклонение» от измененной Х-хромосомы, что приводит к отсутствию заболеваний у женщины-носителя. При преобладании случайной инактивации нормальной Х-хромосомы происходит перекос в сторону мутантного аллеля, и у женщины-носителя может развиться заболевание (хотя и в более легкой форме, чем у мужчин).
• Ген может передаваться от женщины-носителя к ее сыновьям: в семьях с рецессивным заболеванием, сцепленным с Х-хромосомой, часто встречаются больные мужчины, являющиеся родственниками женщин, у которых заболевания нет.
• Мужчины с заболеванием не могут передать его своим сыновьям (при этом все их дочери будут носителями).
Риск того, что у женщины-носителя будет больной ребенок, составляет 25%, иными словами, заболевание может развиться у половины детей мужского пола.
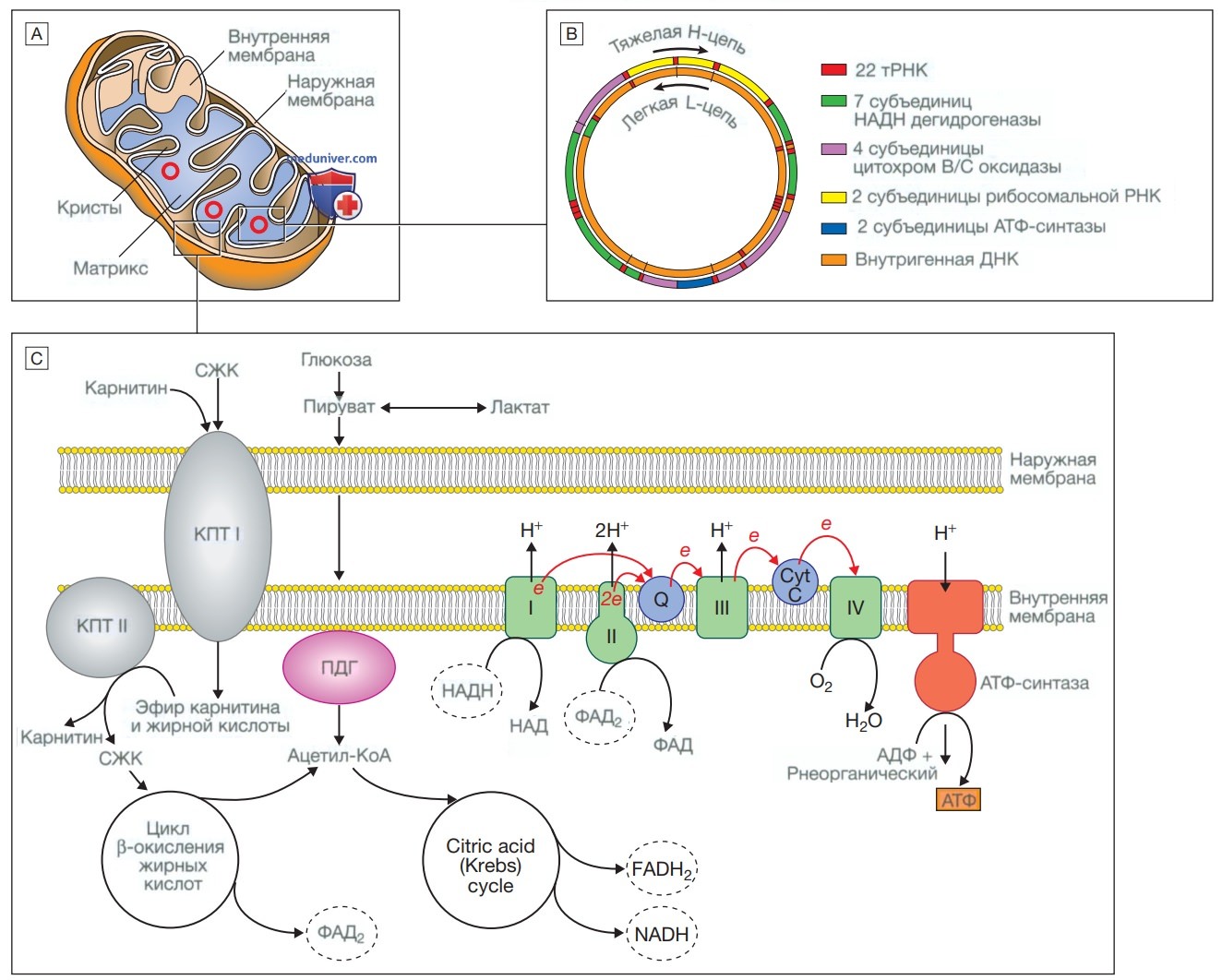
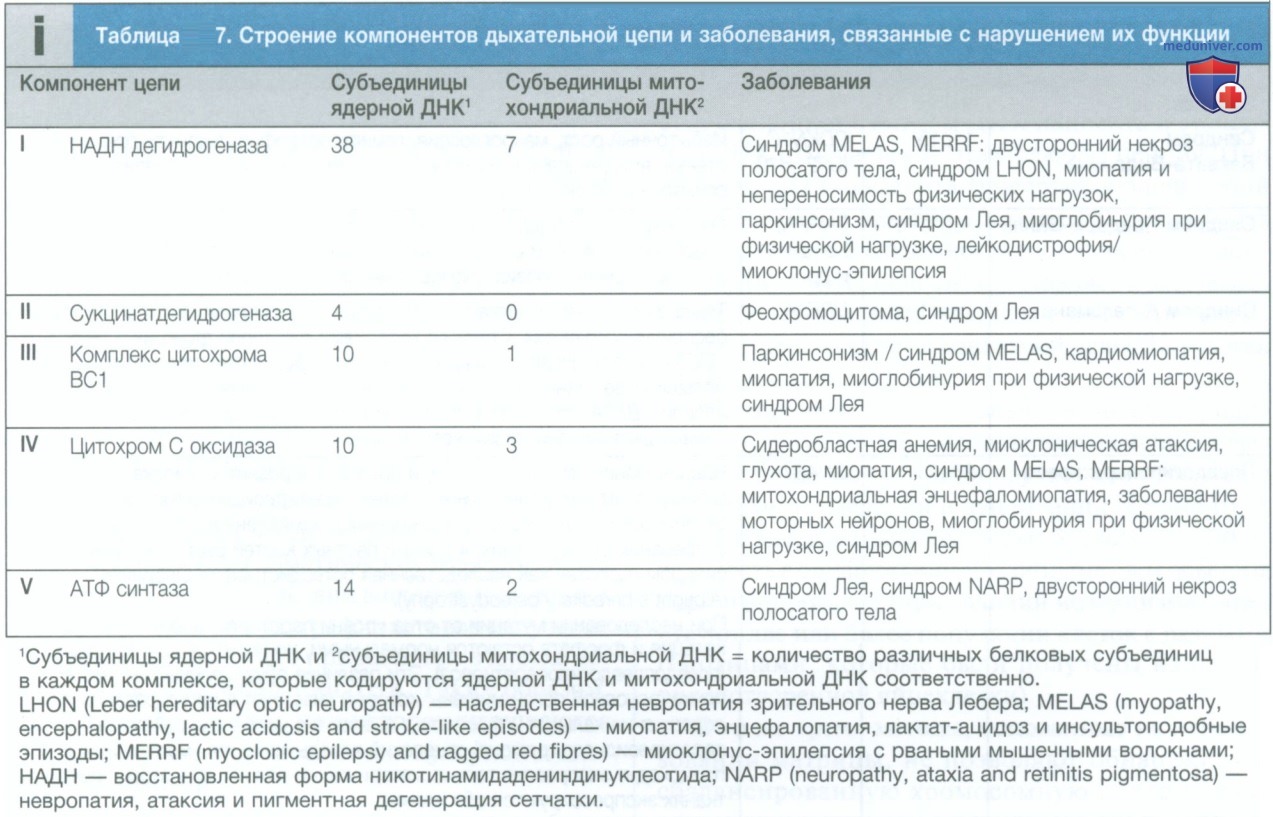
4. Митохондриальное наследование. Митохондрия является основным местом образования энергии в клетке. Митохондрии возникли в процессе эволюции в результате симбиоза с внутриклеточными бактериями. Они имеют характерную структуру с функционально различными внутренней и внешней мембранами. Митохондрии вырабатывают энергию в виде аденозинтрифосфата (АТФ). АТФ в основном образуется в ходе метаболизма глюкозы и жиров (рис. 2). Глюкоза не может напрямую проникать в митохондрии, а сначала превращается в пируват в ходе гликолиза. В дальнейшем пируват переносится в митохондрию и подвергается метаболизму с образованием ацетил-кофермента А (ацетил-КоА).
Рисунок 2. Митохондрия. А — строение митохондрии. Митохондрия имеет две мембраны — гладкую наружную и складчатую внутреннюю, последняя имеет выступы, которые называются кристами. Мембраны формируют два пространства: межмембранное пространство, которое играет важную роль в цепи переноса электронов, и внутреннее пространство (или матрикс), где расположены митохондриальная ДНК и ферменты, участвующие в цикле лимонной кислоты (цикле Кребса) и в цикле β-окисления жирных кислот. В — митохондриальная ДНК. Митохондрия содержит несколько копий кольцевой двухцепочечной ДНК. В ДНК митохондрий есть некодирующий участок и участок, который кодирует гены, отвечающие за образование энергии, молекулы транспортной РНК митохондрий и рибосомальной РНК митохондрий (НАДН — восстановленная форма никотинамидадениндинуклеотида). С — образование энергии в митохондриях. Жирные кислоты переносятся в митохондрию в конъюгированном виде с карнитином, конъюгирование происходит под действием карнитинпальмитилтрансферазы типа 1 (КПТI). Внутри матрикса они расщепляются под действием КПТ II с образованием свободных жирных кислот (СЖК). Последние вступают в цикл β-окисления, в ходе которого образуется ацетил-кофермент А (ацетил-КоА). Пируват может проникать в митохондрию в свободном виде и метаболизируется пируватдегидрогеназой (ПДГ) с образованием ацетил-КоА. Ацетил-КоА включается в цикл Кребса, в результате чего образуются никотинамидадениндинуклеотид и флавинадениндинуклеотид (восстановленная форма). Никотинамидадениндинуклеотид и флавинадениндинуклеотид используются белками в цепи переноса электронов для образования градиента концентрации ионов водорода на внутренней мембране митохондрий. При восстановлении никотинамидадениндинуклеотида и флавинадениндинуклеотида белками I и II соответственно образуются электроны (е), а высвобождающаяся энергия используется для работы протонного насоса с переносом ионов водорода в межмембранное пространство. Кофермент Q10/ убихинон (Q) представляет собой чрезвычайно гидрофобный переносчик электронов, который способен перемещаться в пределах внутренней мембраны. Благодаря переносу электронов белками цепи все большее количество протонов переносится через мембрану до того момента, пока электроны не достигнут комплекса IV (цитохромоксидаза), который использует энергию для восстановления кислорода с образованием молекулы воды. Протонный градиент используется при образовании аденозинтрифосфата под действием фермента аденозинтрифосфат-синтазы, которая состоит из протонного канала и каталитических центров для синтеза аденозиндифосфата из аденозинтрифосфата. При открытии канала ионы водорода попадают в матрикс по градиенту концентрации, при этом происходит выделение энергии, которая используется для образования аденозинтрифосфата
Жирные кислоты переносятся в митохондрии после конъюгации с карнитином и последовательно катаболизируются в результате такого процесса, как бета-окисление с образованием ацетил-КоА. Ацетил-КоА, полученный в результате окисления пирувата и жирных кислот, используется в цикле лимонной кислоты (цикле Кребса). Цикл Кребса представляет собой серию ферментативных реакций, в которых образуется СО2, восстановленная форма никотинамидадениндинуклеотида и восстановленная форма флавинадениндинуклеотида. Затем и никотинамидадениндинуклеотид, и флавинадениндинуклеотид выступают в качестве доноров электронов в дыхательной цепи. Эти электроны передаются в сложной серии реакций, в результате которых на внутренней мембране митохондрии создается протонный градиент. При помощи этого градиента АТФ-синтаза, белок внутренней мембраны митохондрии, образует АТФ, которая затем переносится в другие части клетки.
Дефосфорилирование АТФ используется для получения энергии, необходимой для многих клеточных процессов.
Каждая митохондрия содержит от 2 до 10 копий двухцепочечной кольцевой молекулы ДНК массой 16 килобаз (кБ). Эта митохондриальная ДНК содержит 13 генов, кодирующих белки дыхательной цепи, и гены, некодирующие РНК, необходимые для синтеза белка в митохондриях (см. рис. 2). Частота мутаций митохондриальной ДНК относительно высока из-за отсутствия защиты, которую обеспечивает хроматин. Описаны некоторые заболевания митохондриальной ДНК, характеризующиеся нарушением образования АТФ. Наибольшее количество митохондрий содержится в клетках с высокими метаболическими потребностями, например в клетках мышц, сетчатки и базальных ганглиев.
Как правило, при митохондриальных болезнях эти ткани поражены в наибольшей степени (табл. 7). Существует множество других митохондриальных болезней, которые вызваны мутациями в ядерных генах, кодирующих белки. Эти гены в дальнейшем переносятся в митохондрии и играют важную роль в образовании энергии. Примерами таких заболеваний является большинство форм синдрома Лея (в то же время синдром Лея также может быть вызван мутацией митохондриального гена).
Заболевания, связанные с мутациями митохондриальной ДНК, наследуются от женщин, однако у мужчин и женщин частота их развития, как правило, одинаковая (см. рис. 1). В отличие от других типов наследования, которые упоминались выше, митохондриальное наследование не имеет никакого отношения к мейозу, но отражает тот факт, что передача митохондриальной ДНК осуществляется яйцеклетками: сперматозоиды не участвуют в переносе митохондрий в зиготу. Митохондриальные заболевания, как правило, различаются по пенетрантности и экспрессивности в пределах семьи.
В основном это объясняется тем фактом, что в митохондриях патогенная мутация содержится только в части многочисленных молекул митохондриальной ДНК (степень гетероплазмии мутаций митохондриальной ДНК).
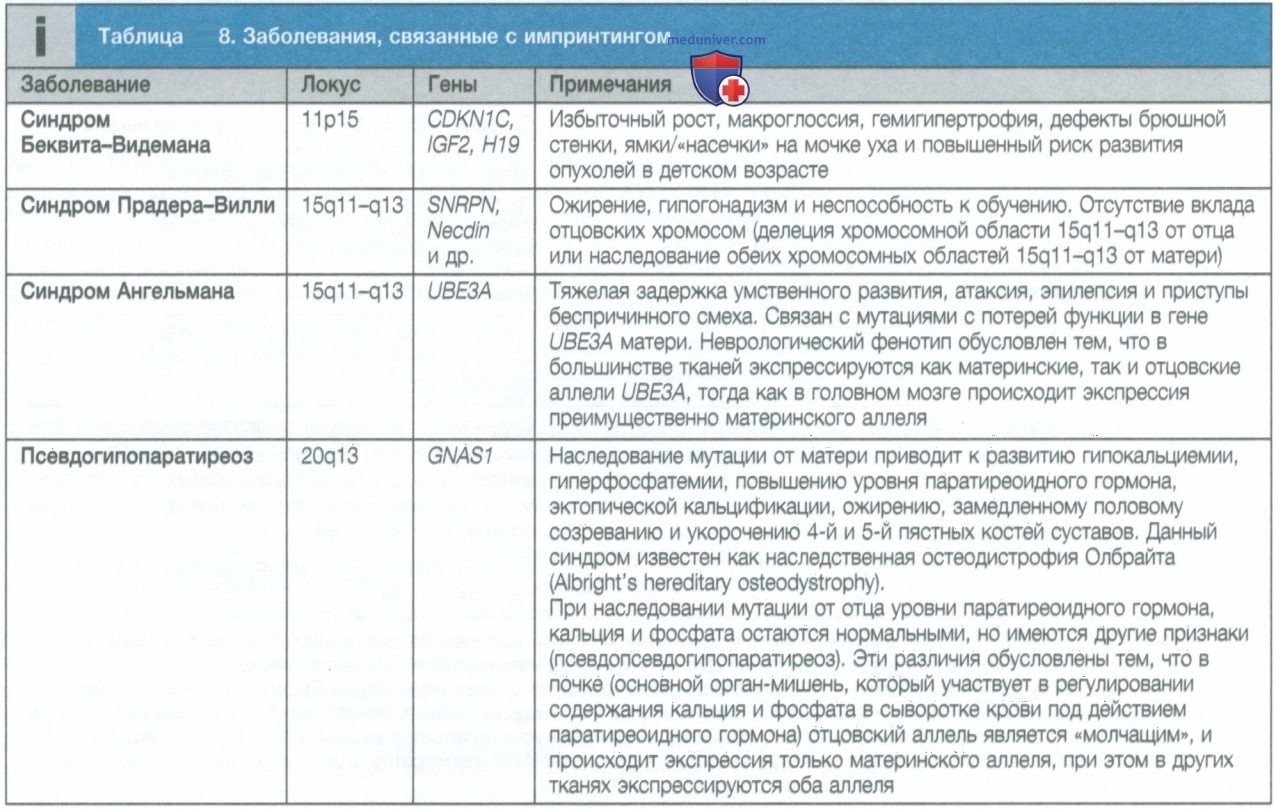
5. Импринтинг. Было идентифицировано несколько участков хромосом (локусов), где экспрессия генов наследуется в зависимости от их родительского происхождения; такие локусы называются локусами импринтинга. В этих локусах может быть активным ген, наследуемый по отцовской линии, тогда как ген, наследуемый по линии матери, может быть «молчащим», или наоборот. В результате мутаций в локусах импринтинга возникает необычный тип наследования, при котором фенотип проявляется только в том случае, если он наследуется от родителя, имеющего транскрипционно активный аллель. Примеры болезней импринтинга приведены в табл. 8.
б) Соматические генетические заболевания. Соматические мутации не наследуются, а возникают уже в ходе митоза клеток после завершения стадии зиготы, начиная с эмбрионального периода и заканчивая пожилым возрастом. Примером таких заболеваний является диссеминированный фиброзный остеит (синдром Мак-Кьюна-Олбрайта), при котором соматическая мутация в гене GS альфа вызывает конститутивную активацию нисходящих сигналов, что приводит к очаговым поражениям скелета и эндокринным нарушениям.
Наиболее важным примером заболеваний у человека, которые обусловлены соматическими мутациями, являются злокачественные новообразования. В данном случае драйверные мутации происходят в генах, участвующих в регуляции деления или апоптоза клеток, в результате чего начинается аномальный рост клеток и возникают опухоли. Основными категориями мутаций, вызывающих рак, являются мутации с приобретением функций в генах, которые стимулируют рост (онкогены), и мутации с потерей функции в генах, которые подавляют рост (гены-онкосупрессоры). Вне зависимости от задействованного механизма для большинства опухолей необходимо наличие активирующей мутации в одной клетке, которая затем может ускользать от контроля нормального роста. Эта клетка начинает чаще реплицироваться или не подвергается запрограммированной гибели, в результате чего развивается экспансия клона.
По мере увеличения размера клона в одной или нескольких клетках могут возникать дополнительные мутации, которые обеспечивают еще большее преимущество в росте. Происходит пролиферация этих субклонов, и в конечном счете может развиться метастатический рак с агрессивным ростом. Благодаря сложному механизму саморегуляции клетки для образования злокачественной опухоли одной мутации обычно недостаточно. Например, если мутация приводит к активации гена или рецептора фактора роста, то в результате аутокринной стимуляции такая клетка будет реплицироваться чаще. Однако данная мутантная клетка по-прежнему будет зависеть от контрольных точек нормального клеточного цикла для обеспечения целостности ДНК в потомстве клеток.
Тем не менее, если в одной и той же клетке возникают дополнительные мутации, приводящие к дефектным контрольным точкам клеточного цикла, происходит быстрое накопление последующих мутаций, которые могут обеспечить полностью нерегулируемый рост, и/или отделение от матрикса и межклеточных контактов, и/или резистентность к апоптозу.
Поскольку рост клеток становится все более и более нерегулируемым, клетки утрачивают способность к дифференцировке, прекращают взаимодействовать с нормальным тканевым окружением и не могут обеспечивать соответствующую сегрегацию хромосом во время митоза. В совокупности эти процессы представляют собой классические характеристики злокачественного неорганизованного роста клеток, которые приводят к различному уровню дифференцировки, а также к аномалиям числа и структуры хромосом. Увеличение частоты соматических мутаций может происходить при воздействии внешних мутагенных факторов, таких как ультрафиолетовое излучение или табачный дым, либо при наличии в клетке дефектов в системе репарации ДНК. Таким образом, рак — это заболевание, при котором нарушаются фундаментальные процессы молекулярной и клеточной биологии.