Многие детские заболевания вызваны мутациями единичных генов, кодирующих определенные белки. Эти мутации могут изменять первичную структуру белка/количество синтезируемого белка. Функция белка, будь то фермент, рецептор, транспортный белок, мембранный компонент, корегулятор транскрипции, структурный элемент, м.б. нарушена/отменена полностью. Наследственные заболевания, которые нарушают нормальные биохим. процессы, называют врожденными нарушениями обмена в-в/наследственными метаболическими заболеваниями.
Большинство генетических изменений являются клинически несущественными и представляют собой доброкачественные вариации. Однако патогенные варианты приводят к заболеваниям, которые могут варьировать по степени тяжести и времени начала. Тяжелые метаболические нарушения обычно проявляются клинически в периоде новорожденности/вскоре после него, тогда как более легкие формы могут проявляться позднее в детстве и даже у взрослых.
За некоторыми исключениями симптомы большинства метаболических состояний не являются специфическими и не позволяют поставить точный диагноз без дальнейшего обследования. Сочетание низкой специфичности симптомов и редкая встречаемость метаболических нарушений затрудняют постановку диагноза. При прогрессировании симптомов, отсутствии данных о негенетическом заболевании после всестороннего обследования, а также наличие аналогичных симптомов в анамнезе у родственников пациента и данных о браке между близкими родственниками педиатру следует проконсультироваться с генетиком и провести метаболическое тестирование на начальных этапах обследования.
Для большинства семей, страдающих метаболическими нарушениями, правильный диагноз зачастую является лишь началом долгого пути лечения. Хотя каждое наследственное метаболическое нарушение в отдельности встречается редко, улучшенная диагностика и увеличение выживаемости пациентов с метаболическими нарушениями фактически гарантируют, что педиатр столкнется с такими пациентами и будет должен оказывать им МП.
Педиатры могут играть ключевую роль в обеспечении непрерывности лечения, регулировании некоторых аспектов лечения, стимулировании пациентов соблюдать предписанный режим, а также в проведении рутинных педиатрических вмешательств, таких как вакцинация, направление к специалистам и генетическое консультирование.
Повышение осведомленности о метаболических состояниях, более широкая доступность биохим. лабораторий, общий метаболомный анализ и рутинное применение секвенирования экзома резко повысили уровень выявления известных нарушений и способствовали обнаружению новых метаболических заболеваний. При этом сбор и анализ семейного анамнеза остается важнейшим скрининговым тестом, который медработник может использовать для выявления риска метаболического нарушения у младенцев/детей. Значение для дальнейших исследований может иметь идентификация кровного родства/особого этнического происхождения с необычно высокой частотой врожденных нарушений метаболизма.
Напр., тирозинемия 1-го типа чаще встречается у канадцев французского происхождения из г. Квебек, болезнь «кленового сиропа» чаще встречается у амишей в США, а болезнь Канавана (Canavan) — у евреев ашкенази.
а) Скрининг новорожденных. Редкость отдельно рассматриваемых врожденных метаболических нарушений, важность ранней диагностики и последующая необходимость генетических консультаций являются сильным аргументом в пользу всеобщего скрининга всех новорожденных. Основу скрининга новорожденных в настоящее время составляют тандемная масс-спектрометрия метаболитов и цифровой микро-флюидный анализ активности ферментов. Для выполнения этих методов необходимо поместить несколько капель крови на фильтровальную бумагу и доставить образцы в центральную лабораторию для анализа.
Указанные методы позволяют идентифицировать многие генетические нарушения, при этом перечень заболеваний продолжает увеличиваться (табл. 1, 2). Педиатрам необходимо знать общие аспекты методики и ограничения скрининга. При «+» результате скрининга может потребоваться повторный скрининг новорожденного/тестирование для подтверждения диагноза. Время, необходимое для получения результатов после отправки образцов, может отличаться в разных странах и даже в разных регионах одной страны.
Некоторые метаболические состояния м.б. достаточно серьезными и клинически манифестируют до получения результатов скрининга новорожденных. И наоборот, метаболиты, используемые для диагностики нарушений, включенных в скрининг, при более легких формах могут не достигать установленного порога для проведения повторных исследований, что приводит к «-» результатам скрининга новорожденных и задержке диагностики. Поэтому «-» результат скрининга новорожденного с симптомами, предполагающими метаболическое нарушение, требует направления в центр генетики для дальнейшего обследования.
Универсальный скрининг новорожденных также может выявить легкие формы наследственных метаболических состояний, некоторые из них могут никогда не вызвать клинических проявлений в течение жизни человека. Напр., в программах скрининга с использованием тандемной масс-спектрометрии неожиданно часто встречался дефицит короткоцепочечной ацил-КоА-дегидрогеназы, но большинство этих детей так и оставались бессимптомными. Это подчеркивает необходимость постоянной оценки пороговых значений метаболитов и подходов к подтверждающему тестированию для достижения максимального диагностического результата и минимизации потенциальных психосоциальных и экономических последствий таких результатов. Недоношенные дети являются особой группой пациентов, у которых частота л/пол/о результатов тестов м.б. особенно высокой.
С появлением генетической терапии СМА и ферментозамещающей терапии некоторых лизосомных болезней накопления (напр., болезни Помпе (Pompe), болезни Фабри (Fabry), болезни Гоше (Gaucher) и мукополисахаридоза типа 1) пилотные программы скрининга новорожденных на первоначальном этапе успешно выявляли СМА/лизосомные болезни накопления, часто до развития тяжелых симптомов.
б) Клинические проявления генетических метаболических заболеваний. Врачам и др. медработникам, оказывающим помощь детям, следует ознакомиться с ранними проявлениями генетических нарушений метаболизма, поскольку 1) симптомы тяжелых форм некоторых из этих состояний могут проявиться до получения результатов скрининговых исследований, 2) существующие в настоящее время скрининговые методы, хотя и являются довольно обширными, выявляют небольшое количество всех наследственных метаболических состояний. В неонатальный период клинические проявления обычно являются неспецифичными и аналогичны симптомам сепсиса у младенцев.
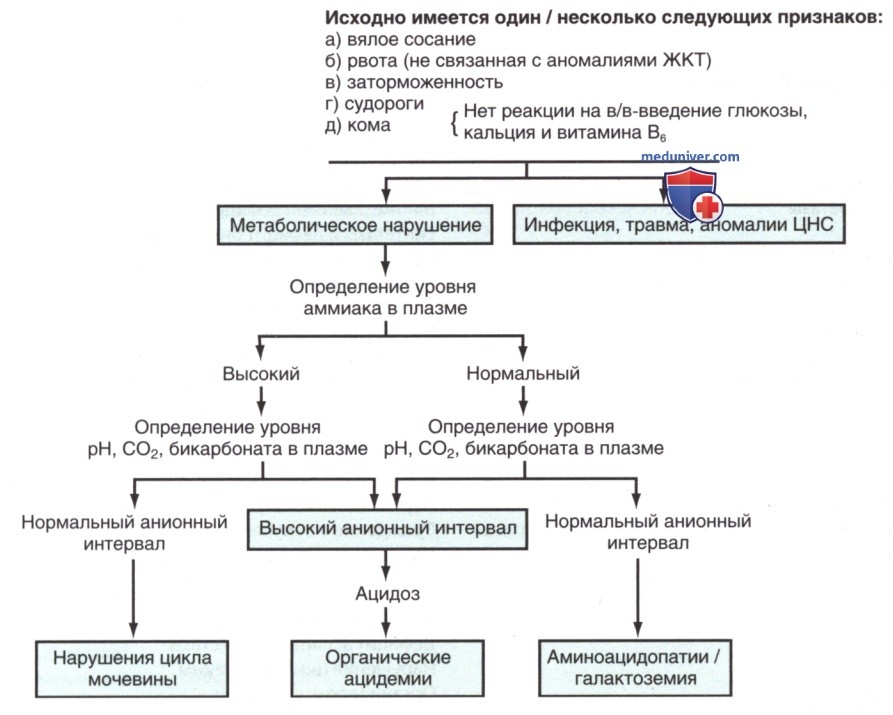
При ДД тяжелобольного новорожденного необходимо учитывать возможность генетического нарушения метаболизма, а в группах высокого риска следует провести специальные исследования (рис. ниже).
Первичный клинический подход к доношенному новорожденному с подозрением на генетическое нарушение метаболизма. Данная схема является руководством для выявления некоторых метаболических нарушений у новорожденных. Схема, хотя и имеет некоторые исключения, подходит для большинства случаев нарушенного/промежуточного метаболизма
Объективные и субъективные симптомы, такие как вялость, гипотония, гипотермия, судороги (табл. 3), вялое сосание и рвота, могут развиться уже через несколько часов после рождения. В некоторых случаях рвота бывает настолько сильной, что возникает подозрение на пилоростеноз, которого обычно нет (хотя у таких младенцев он может наблюдаться в качестве сопутствующего заболевания). Заторможенность, вялое сосание, судороги и кома также могут наблюдаться у младенцев с гипогликемией (табл. 4), гипокальциемией и гипераммониемией (табл. 5). В постановке этих диагнозов помогают измерения концентрации глюкозы и кальция в крови и реакция на в/в введение декстрозы («Глюкозы»)/ кальция. Метаболические нарушения могут затронуть любые органы и системы органов.
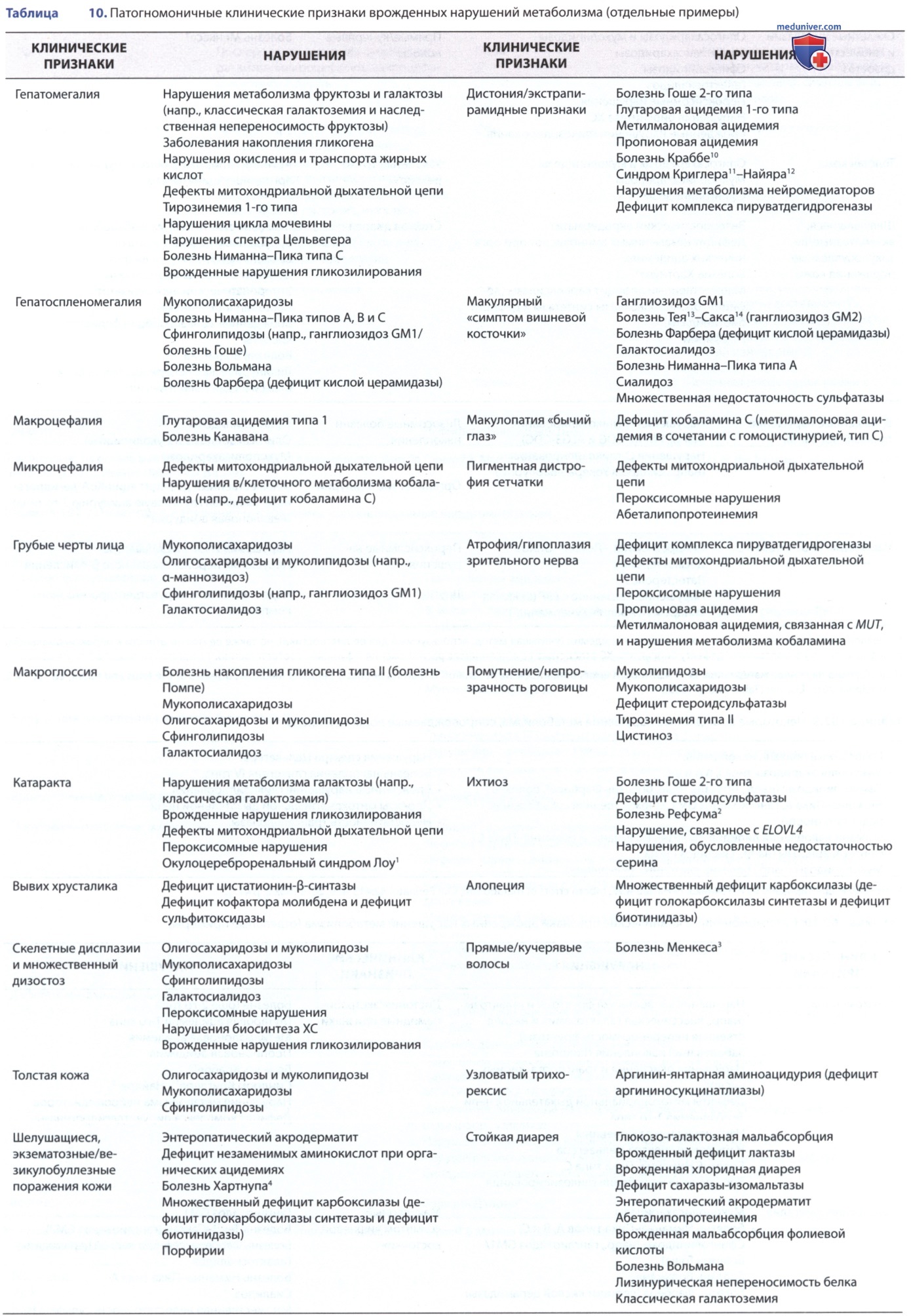
Однако физикальный осмотр обычно выявляет неспецифические проявления; большинство симптомов обусловлены нарушениями со стороны ЦНС, напр. при болезни «кленового сиропа» (лейциноз — прим. науч. ред.) наблюдается опистотонус. При многих врожденных метаболических нарушениях часто наблюдаются гепатомегалия (табл. 6). Кардиомиопатия (табл. 7), дисморфические особенности (табл. 8) и водянка плода (табл. 9) являются дополнительными возможными проявлениями метаболического нарушения (табл. 10). Иногда большую помощь в диагностике может оказать специфический запах (табл. 11).
У все большего числа пациентов выявляют нарушение метаболизма через несколько месяцев/лет после рождения. Это характерно для пациентов с более легкими АуР-патогенными вариантами, при митохондриальных нарушениях, у девочек с Х-сцепленными рецессивными состояниями и при специфических метаболических состояниях, которые обычно возникают в более позднем возрасте. Наследственное нарушение метаболизма у новорожденных можно предположить при таких клинических проявлениях, как задержка умственного развития, нарушение двигательной функции, регресс развития, судороги, психоз, кардиомиопатия, миопатия, органомегалия и рецидивирующая рвота (табл. 12). Может наблюдаться эпизодическая/прерывистая картина с эпизодами острых клинических проявлений, разделенных периодами, казалось бы, безрецидивных состояний, когда дети кажутся здоровыми.
Приступы обычно провоцируются стрессами/неспецифическими катаболическими процессами, вызванными, напр., инфекцией. В период такого острого приступа ребенок может умереть. Обследование на врожденные дефекты метаболизма должно проводиться у ребенка, имеющего одно/несколько из следующих проявлений: необъяснимая задержка в развитии, задержка умственного развития; регресс психического развития, нарушение двигательных функций/навязчивые движения (напр., дистония, хореоатетоз, атаксия), судороги, кататония, необычный запах (особенно при остром заболевании), периодические эпизоды необъяснимой рвоты, ацидоза, психического нарушения, психоза/комы, гепатомегалия, камни в почках, нарушение функции почек, особенно синдром Фанкони (Fanconi)/почечноканальцевый ацидоз, мышечная слабость и кардиомиопатия (табл. 12).
Для диагностики обычно требуется проведение ряда специфических лабораторных исследований. В качестве стартовых скрининговых тестов для диагностики предполагаемого врожденного нарушения метаболизма полезно определить уровни аминокислот и профиль ацилкарнитина в плазме, профиль общего и свободного карнитина и уровень органических кислот в моче, хотя диагностические возможности этих показателей не являются исчерпывающими. Анализ содержания аммиака, лактатов, бикарбонатов и pH в плазме доступны в стационаре и очень информативны на начальном этапе при проведении ДД основных причин генетических нарушений метаболизма (табл. 13; см. рис. выше). Повышение уровня аммиака в крови обычно вызвано дефектами ферментов цикла мочевины, органическими ацидемиями и нарушениями окисления жирных кислот.
Новорожденные с повышенным уровнем аммиака в крови из-за нарушения цикла мочевины, как правило, имеют нормальные значения pH и бикарбонатов в сыворотке; без измерения аммиака в крови их заболевание м.б. не диагностировано, что может привести к летальному исходу. При органических ацидемиях повышенный уровень аммиака в плазме сопровождается тяжелым ацидозом, вызванным накоплением органических кислот, кетоновых тел и лактата в жидкостях организма.
При нормальных уровнях аммиака, pH и бикарбонатов в крови следует рассматривать др. аминоацидопатии (напр., гиперглицинемию)/галактоземию. Галактоземия у новорожденных может также манифестировать в виде катаракты, гепатомегалии, асцита и желтухи.
в) Лечение. Большинство пациентов с генетическими нарушениями метаболизма реагируют на один/несколько следующих методов лечения.
• Большое значение в лечении больных детей имеют специальные диеты. Диеты должны быть адаптированы к патофизиологии состояния и значительно отличаются в зависимости от заболевания.
• Для быстрого удаления накопившихся токсичных соединений применяется гемодиализ. Это очень эффективный метод, используемый в острую фазу заболевания.
• При лечении катаболических состояний у пациентов с риском метаболического кризиса можно применять р-ры, содержащие декстрозу («Глюкозу») и электролиты.
• Введение дефицитного метаболита.
• Введение кофактора/кофермента для максимального увеличения остаточной активности фермента.
• Активация альтернативных путей для уменьшения количества токсичных соединений, накапливаемых вследствие генетической мутации.
• Введение дефицитного фермента.
• Трансплантация костного мозга.
• Трансплантация печени и почек.
Трансплантация органов м.б. наилучшим методом лечения для стабилизации пациента с метаболическими нарушениями и улучшения качества его жизни. В настоящее время замена мутантного гена нормальной копией с помощью генной терапии была успешной только при некоторых заболеваниях.
Лечение генетических нарушений обмена в-в сложное и требует мед. и технических знаний. Часто необходимо адаптировать схему лечения к конкретному пациенту, поскольку фенотипические различия тяжести заболевания м.б. значительными даже в одной семье. Обучение и поддержка семьи является ключом к успешной долгосрочной терапии. Даже если пациент имеет неблагоприятный прогноз, необходимо приложить все усилия для постановки правильного диагноза до наступления смертельного исхода. Наибольший эффект от терапии достигается при работе группы специалистов (специалиста по метаболической генетике, диетолога, невролога и психолога) в крупном мед. центре.