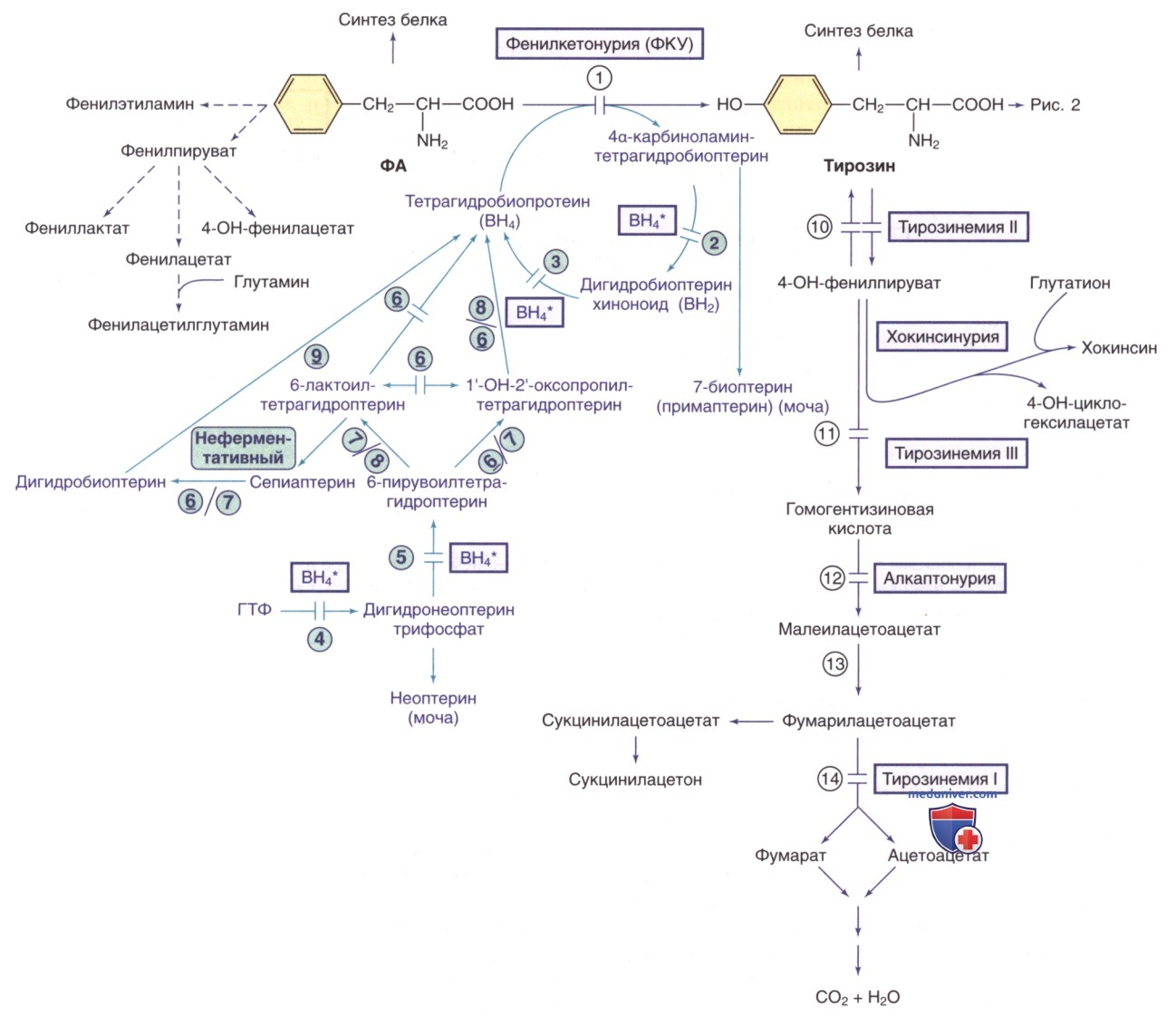
Тирозин выделяется из белков, поступающих с пищей/синтезируется эндогенно из ФА. Это необходимый компонент при синтезе белков и предшественник дофамина, норэпинефрина, адреналина, меланина и тироксина. Избыток тирозина расщепляется до диоксида углерода и воды (см. рис. 1). К наследственным причинам гипертирозинемии относятся дефицит ферментов фумарилацетоацетатгидролазы, тирозинаминотрансферазы и 4-гидроксифенилпируватдиоксигеназы. Приобретенная гипертирозинемия может возникать при тяжелой гепатоцеллюлярной дисфункции (печеночная недостаточность), цинге (витамин С является кофактором 4-гидроксифенилпируватдиоксигеназы) и гипертиреозе.
Рисунок 1. Пути метаболизма фенилаланина (ФА) и тирозина. Генетически детерминированные ферментативные нарушения показаны горизонтальными линиями, пересекающими стрелку(-и) реакции. Пути синтеза кофактора тетрагидробиоптерина (ВН4) показаны фиолетовым цветом. ВН4* относится к дефектам метаболизма ВН4, влияющим на гидроксилазы ФА, тирозина и триптофана (рис. 2 и 5). Ферменты: 1) фенилаланингидроксилаза; 2) птеринкарбиноламиндегидратаза; 3) дигидробиоптеринредуктаза; 4) гуанозинтрифосфат (ГТФ)-циклогидролаза; 5) 6-пирувоилтетрагидроптеринсинтаза; 6) сепиаптеринредуктаза; 7) карбонилредуктаза; 8) альдолазоредуктаза; 9) дигидрофолатредуктаза; 10) тирозинаминотрансфераза; 11) 4-гидроксифенилпируватдиоксигеназа; 12) диоксигеназа гомогентизиновой кислоты; 13) малеилацетоацетат изомераза; 14) фумарилацетоацетат гидролаза
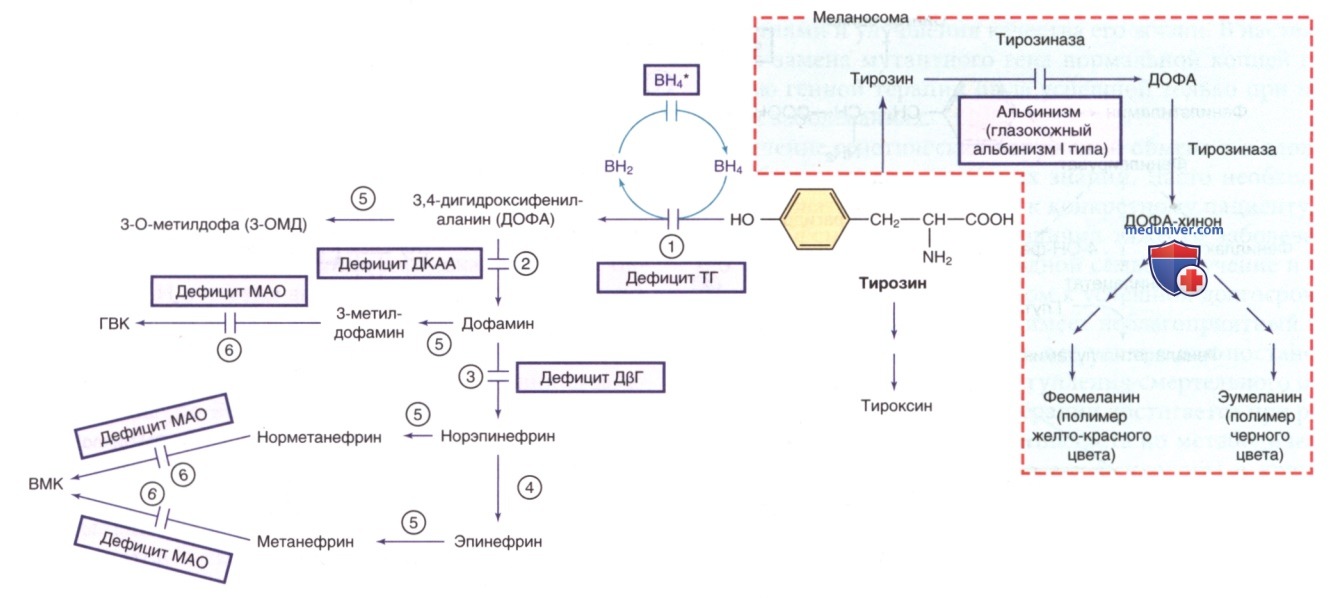
Рисунок 2. Другие пути метаболизма тирозина. ВН4* указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). ГВК — гомованиловая кислота; ВМК — ванилилминдальная кислота; ВН4 — тетрагидробиоптерин. Ферменты: 1) тирозингидроксилаза (ТГ); 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) дофамин-β-гидроксилаза (ДβГ); 4) фенилэтаноламин-N-метилтрансфераза (ФНМТ); 5) катехол-О-метилтрансфераза (КОМТ); 6) МАО
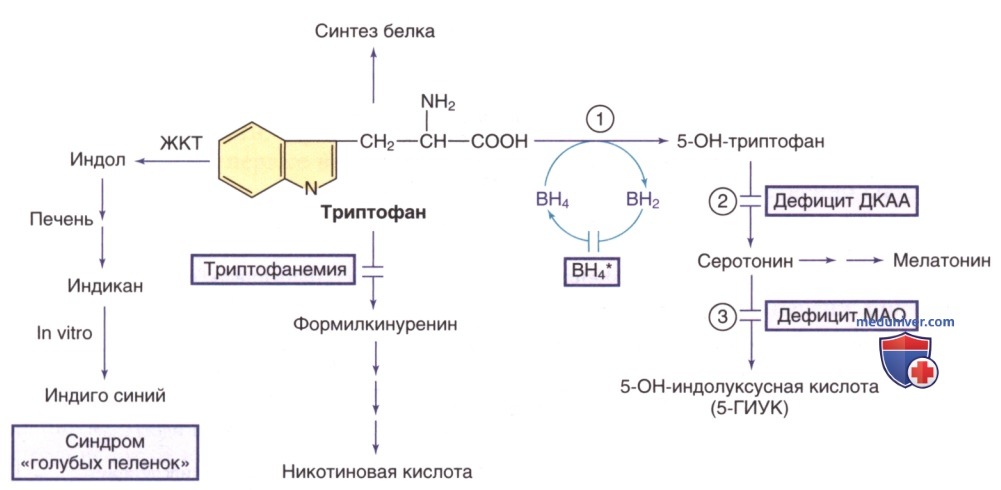
Рисунок 5. Пути метаболизма триптофана. Тетрагидробиоптерин (ВН4*) указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). Ферменты: 1) триптофангидроксилаза; 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) МАО
Повышение тирозина в сыворотке крови может наблюдаться сразу после приема пищи, а также у недоношенных детей.
а) Тирозинемия I типа (дефицит фумарилацетоцетатгидролазы, гепаторенальная тирозинемия). Тирозинемия I типа, вызванная дефицитом фумарилацетоацетатгидролазы, является тяжелой полиорганной патологией. Метаболиты тирозина, особенно фумарилацетоацетата и сукцинилацетона, вызывают поражение печени, почек и нервов.
1. Клиническая картина и патогенез заболевания. Новорожденные с тирозинемией I типа, не получающие лечение, выглядят при рождении как обычные дети, а симптомы возникают в первый год жизни. У большинства пациентов симптоматика проявляется в 2-6 мес, но в редких случаях симптомы могут манифестировать в первый месяц, или, напротив, ребенок может казаться здоровым в первый год жизни. Более раннее проявление заболевания имеет неблагоприятный прогноз. У младенцев, не получающих лечение, с манифестацией симптомов <2 мес смертность в течение первого года жизни составляет 60%, тогда как у младенцев с дебютом заболевания >6 мес показатель смертности снижается до 4%.
Типичным предвестником болезни является печеночный криз, спровоцированный интеркуррентным заболеванием, вызвавшим нарушение катаболизма. Часто наблюдаются лихорадка, повышенная возбудимость, рвота, кровотечение, гепатомегалия, желтуха, гипертрансаминаземия, гипогликемия и невропатия. Следствием повышенного содержания метаболитов метионина является появление запаха, напоминающего вареную капусту. Печеночные кризы способны прогрессировать в печеночную недостаточность и приводить к летальному исходу. В периодах между кризами сохраняются задержка физического развития разл. степени выраженности, гепатомегалия и нарушения коагуляции. С возрастом развивается цирроз и, наконец, гепатоцеллюлярная карцинома.
У 40% детей с такой патологией наблюдаются эпизоды острой периферической невропатии, напоминающей острую порфирию. Такие кризы часто спровоцированы незначительной инфекцией; характеризуются сильной болью, чаще в ногах, сопровождающейся гипертонусом разгибательных мышц шеи и туловища; рвотой, паралитической непроходимостью кишечника, а иногда и самопроизвольными травмами языка/слизистой оболочки щек. В 30% случаев наблюдается выраженная гипотония, которая может привести к ДН, требующей ИВЛ. Кризы обычно длятся 1-7 дней, но восстановление после паралитического криза может занимать недели/месяцы.
Поражение почек проявляется в виде Фанкони (Fanconi) - подобного синдрома с гиперфосфатурией, гипофосфатемией, метаболическим ацидозом с нормальной анионной разностью и витамин-D-резистентным рахитом. При УЗИ м.б. выявлены нефромегалия и нефрокальциноз. У подростков и взрослых пациентов может развиться почечная недостаточность. У некоторых младенцев наблюдаются ГКМП и гиперинсулинизм.
2. Лабораторные данные. ДК тирозинемии I типа является повышенное содержание сукцинилацетона в сыворотке крови и моче (см. рис. 1). У пациентов, получавших нитизинон, содержание сукцинилацетона может опуститься ниже диагностического порога. У пациентов, не получавших лечение, часто значительно повышено содержание АФП в крови, у большинства пациентов снижены факторы свертывания крови, синтезируемые печенью. У новорожденных с данной патологией в пуповинной крови повышено содержание АФП, что указывает на в/утробное поражение печени. Часто повышается содержание печеночных трансаминаз, причем значительное повышение отмечается при острых печеночных кризах. Концентрация билирубина в сыворотке крови обычно соответствует норме, но при печеночной недостаточности этот показатель может повышаться. В момент постановки диагноза обычно наблюдаются повышенные уровни тирозина в плазме, что, однако, неспецифично и зависит от приема пищи.
У пациентов с поражением печени содержание др. аминокислот в сыворотке крови, особенно метионина, также м.б. повышенным. Возможно появление гиперфосфатурии, гипофосфатемии и генерализованной аминоацидурии. В моче повышается содержание 5-аминолевулиновой кислоты (др. название — δ-аминолевулиновая кислота) вследствие ингибирования 5-аминолевулинатдегидратазы сукцинилацетоном (см. рис. 1).
Диагностика обычно основывается на выявлении повышенного уровня сукцинилацетона в моче/крови.
Неонатальный скрининг на гипертирозинемию с использованием только тирозина выявляет лишь часть пациентов с тирозинемией I типа. Сукцинилацетон, определение которого включено во многие программы неонатального скрининга, имеет более высокие ЧС, чем тирозин, и является предпочтительным метаболитом для скрининга*.
P.S. * КР РФ по оказанию МП детям с наследственной тирозинемией 1-го типа. Баранов А.А., Намазова-Баранова Л.С., Куцев С.И. и др., 2015 г.
Следует отличать тирозинемию I типа от др. причин гепатита и печеночной недостаточности у младенцев, в т.ч. от галактоземии, наследственной непереносимости фруктозы, неонатального гемохроматоза, неонатального гепатита/гигантоклеточного гепатита и цитруллинемии II типа.
3. Лечение и исход. Диета с низким содержанием ФА и тирозина позволяет замедлить, но не остановить прогрессирование заболевания. ЛП выбора является нитизинон, который ингибирует 4-гидроксифенилпируватдиоксигеназу и снижает поступление метаболитов тирозина к фумарилацетоацетатгидролазе, тем самым уменьшая синтез соединений (фумарилацетоацетата и сукцинилацетона), являющихся причиной заболевания (см. рис. 1). Дозу нитизинона титруют до самой низкой и наиболее эффективной дозы (обычно целевым диапазоном в сыворотке крови является 20-40 мкмоль/л) для подавления синтеза сукцинилацетона при поддержании уровня тирозина в плазме <400 мкмоль/л (7,2 мг/дл). Такое лечение предотвращает острые печеночные и неврологические кризы. Хотя нитизинон значительно замедляет прогрессирование заболевания, повреждение печени, возникшее до начала лечения, является необратимым. Поэтому пациентов необходимо наблюдать на предмет развития цирроза/гепатоцеппюлярной карциномы. При визуализации наличие даже одного узла в печени обычно указывает на цирроз.
У пациентов с тирозинемией большинство узлов в печени являются доброкачественными, но современные методы визуализации не позволяют достоверно выявить злокачественные узлы. Для пациентов с тяжелой печеночной недостаточностью, не отвечающих на лечение нитизиноном, эффективным методом лечения является трансплантация печени, что также позволяет снизить риск развития гепатоцеллюлярной карциномы. Влияние лечения нитизиноном на необходимость трансплантации печени все еще находится на стадии исследования, но наибольший эффект наблюдается у пациентов, получивших лечение на ранней стадии заболевания, до развития клинических симптомов, напр. у детей, выявленных при неонатальном скрининге. У пациентов, начавших лечение на ранней стадии, нитизинон в значительной степени снижает необходимость трансплантации печени. Поскольку данный ЛП повышает содержание тирозина в плазме, рекомендуется соблюдать диету с низким содержанием тирозина и ФА.
В редких случаях у пациентов, принимающих нитизинон, в роговице появляются кристаллы, предположительно тирозина, но при строгом соблюдении диеты это явление обратимо. Данное нарушение в сочетании с задержкой развития у некоторых пациентов с тирозинемией II типа и сохраняющимся повышенным уровнем тирозина указывает на необходимость продолжения соблюдения диеты с низким содержанием ФА и тирозина у пациентов, принимающих нитизинон. При диетотерапии с ограничением тирозина и ФА необходимо оценивать динамику роста и развития, при этом обеспечивая достаточное потребление аминокислот и др. питательных в-в.
4. Генетика и распространенность. Тирозинемия I типа наследуется по АуР-типу. Ген FAH расположен в хромосоме 15q25.1. Пренатальное молекулярно-генетическое исследование показано в случаях отягощенного семейного анамнеза, а также для определения носителей конкретных мутаций в группах риска, напр. среди франко-канадцев из региона Сагеней-Лак-Сен-Жан в г. Квебек. В этом регионе данное заболевание встречается в 1:1846 живорожденных, тогда как мировая статистика составляет 1:100 000 живорожденных. При пренатальном скрининге измеряют содержание сукцинилацетона в околоплодных водах. Пренатальная диагностика при отягощенном наследственном анамнезе по тирозинемии возможна методом анализа ДНК амниоцитов/ворсинок хориона.
б) Тирозинемия II типа (дефицит тирозинаминотрансферазы, синдром Рихнера-Ханхарта (Richner-Hanhart), кожно-глазная тирозинемия). Тирозинемия II типа представляет собой редкое АуР-заболевание, вызванное дефицитом цитозольной тирозинаминотрансферазы и приводящее к ладонному и подошвенному гиперкератозу, герпетическим язвам роговицы и умственной отсталости (см. рис. 1). В 6 мес может наблюдаться манифестация глазного синдрома, в т.ч. чрезмерное слезотечение, покраснение, боль и светобоязнь. Предполагается, что поражение роговицы вызвано отложением тирозина. В отличие от герпетических язв, при тирозинемии II типа поражения роговицы плохо окрашиваются флуоресцеином и часто являются двусторонними. В более позднем возрасте могут развиться поражения кожи, в т.ч. болезненные, не зудящие гиперкератотические бляшки на ступнях, ладонях и кончиках пальцев. У ~50% пациентов наблюдается умственная отсталость легкой/средней степени. Роль близкородственных браков в этом редком заболевании до конца не изучена.
Основным лабораторным признаком у пациентов, не получавших лечение, является выраженная гипертирози-немия (>500 мкмоль/л), которая может достигать 1100-2750 мкмоль/л. Как ни странно, содержание 4-гидрокси-фенилпировиноградной кислоты и ее метаболитов в моче также повышено, хотя эти показатели находятся ниже метаболического блока (см. рис. 1). Выдвинута гипотеза, что это связано с воздействием др. трансаминаз в присутствии высоких концентраций тирозина; синтезируется 4-гидроксифенилпировиноградная кислота в митохондриях, где она не может подвергаться дальнейшему расщеплению. В отличие от тирозинемии I типа, печень и почки функционируют нормально, при этом концентрации др. аминокислот и сукцинилацетона в сыворотке крови остаются в пределах нормы. Тирозинемия II типа развивается в результате патологии гена тирозинаминотрансферазы, обусловливающей дефицит ее цитозольной активности в печени. Этот ген расположен в хромосоме 16q22.
У пациентов с клиническими проявлениями, указывающими на тирозинемию II типа, диагноз подтверждается определением концентрации тирозина в плазме. Возможно проведение молекулярной диагностики. Для определения активности тирозинаминотрансферазы в печени необходима биопсия, поэтому этот показатель определяют редко.
Лечение диетой с низким содержанием тирозина и ФА направлено на поддержание уровня тирозина в плазме <500 мкмоль/л, что уменьшает кожные и глазные проявления. Раннее начало диетотерапии позволяет предотвратить умственную отсталость, что было продемонстрировано в нескольких клинических случаях.
в) Тирозинемия III типа (первичный дефицит 4-гидроксифенилпируватдиоксигеназы). Зарегистрировано всего несколько случаев пациентов с тирозинемией III типа. Большинство из них были выявлены с помощью аминокислотной хроматографии, используемой при разл. неврологических симптомах; и такое выборочное наблюдение, вероятно, искажает наше современное понимание этой патологии. По-видимому, бессимптомные пациенты с дефицитом 4-гидроксифенилпируватдиоксигеназы выявляются при неонатальном скрининге на гипертирозинемию. Возраст пациентов на момент обращения составлял 1-17 мес. У пациентов с явными симптомами зарегистрированы задержка психического развития, судороги, периодическая атаксия и самоповреждающее поведение. Аномалии печени и почек отсутствуют.
Роль дефицита 4-гидроксифенилпируватдиоксигеназы в механизмах заболевания требует дальнейшего изучения. Подозрение в отношении этого диагноза возникает, если у ребенка наблюдается устойчивое умеренное повышение уровня тирозина в плазме (в основном 350-700 мкмоль/л при обычном питании), а в моче присутствует 4-гидроксифенилпировиноградная кислота и ее метаболиты (4-гидроксифенилмолочная и 4-гидроксифенилуксусная кислоты). Диагноз уточняется по наличию патогенных вариантов в гене HPD, кодирующем 4-гидроксифенилпируватдиоксигеназу/в редких случаях по низкой активности фермента 4-гидроксифенилпируватдиоксигеназа; последний вариант требует биопсии печени и обычно не проводится.
Учитывая возможные неврологические нарушения, диета, направленная на снижение уровня тирозина в плазме, является разумной мерой. Также целесообразно определить уровень витамина С (кофактора 4-гидроксифенилпируватдиоксигеназы) в сыворотке крови. Заболевание наследуется по АуР-типу.
г) Хокинсинурия. Миссенс-мутация c.722A>G (p.Asn241Ser) в HPD, кодирующем 4-гидроксифенилпируватдиоксигеназу, приводит к разобщению нормального окисления 4-ги-дроксифенилпирувата до гомогентизиновой кислоты и преждевременному высвобождению хинолуксусной кислоты. Дефектный фермент, неспособный нормально окислять 4-гидроксифенилпируват до гомогентизиновой кислоты, образует промежуточное соединение, которое реагирует с глутатионом с образованием необычной органической кислоты хокинсин [(2-L-цистеин-S-ил-1,4-дигидроксициклогекс-5-ен-1-ил) уксусная кислота], названной по фамилии первого пациента (см. рис. 1). Хокинсинурия может привести к вторичному дефициту глутатиона. Хокинсинурия наследуется по АуД-типу. У одного пациента сложная гетерозиготность по аллелям хокинсурии и тирозинемии типа III привела только к биохим. признакам хокинсурии.
Клиническое течение этого редкого заболевания до конца не изучено. У пациентов с хокинсинурией симптомы проявляются только в младенчестве. Обычно они появляются в первые несколько месяцев жизни, часто после прекращения грудного вскармливания и введения продуктов с высоким содержанием белка. Было отмечено проявление данного заболевания в виде тяжелого метаболического ацидоза, кетоза, задержки физического развития, анемии, умеренной гепатомегалии, почечного канальцевого ацидоза и необычного запаха. Психомоторное развитие обычно не нарушено.
У младенцев с клинической манифестацией, а также у бессимптомных детей и взрослых в моче наблюдается экскреция хокинсина (4-гидроксифенилпировиноградной кислоты) и ее метаболитов (4-гидроксифенилмолочной и 4-гидроксифенилуксусной кислот), 4-гидроксицикло-гексилуксусной кислоты и 5-оксопролина (в результате вторичного дефицита глутатиона). Содержание тирозина в плазме умеренно повышено у детей с клиническими проявлениями, но может оказаться нормальным у бессимптомных пациентов.
Лечение заключается в соблюдении низкобелковой диеты в младенчестве. Приветствуется грудное вскармливание. Важно избегать чрезмерного ограничения белка, поскольку у некоторых пациентов дефицит белка может вызывать задержку физического развития. Сообщалось, что длительное применение N-ацетил-L-цистеина для лечения вторичного дефицита глутатиона было успешным. Рекомендуется контроль уровня витамина С в крови. Дефектный фермент чувствителен к ингибированию нитизиноном. Клинические исследования, показывающие эффективность данного ЛП у младенцев с клиническими проявлениями, не проводились, и показания к его применению не разработаны.
д) Транзиторная тирозинемия у новорожденного. У небольшого числа новорожденных в первые 2 нед жизни уровень тирозина в плазме может достигать 3300 мкмоль/л. Большинство новорожденных с данной патологией являются недоношенными и получают диету с высоким содержанием белка. Считается, что транзиторная тирозинемия развивается в результате задержки созревания 4-гидроксифенилпируватдиоксигеназы (см. рис. 1). У некоторых пациентов отмечаются вялость, нарушения вскармливания и снижение двигательной активности. В большинстве случаев заболевание протекает бессимптомно и выявляется благодаря высокому уровню ФА/тирозина в крови в результате обычного скрининга.
К лабораторным признакам относятся значительное повышение тирозина и умеренное увеличение ФА в плазме. Это отличает гипертирозинемию от ФКУ В моче присутствуют 4-гидроксифенилпировиноградная кислота и ее метаболиты. Гипертирозинемия обычно проходит самостоятельно в первые 2 мес жизни. Это состояние можно скорректировать, уменьшив количество белка, поступающего с пищей, до уровня <2 г/кг в сутки и назначив аскорбиновую кислоту («Витамин С»). У некоторых младенцев с таким заболеванием наблюдалась легкая форма умственной отсталости, но этиологическая связь с гипертирозинемией окончательно не установлена.
е) Алкаптонурия. Алкаптонурия — это редкое (1:250 000 живорожденных) АуР-заболевание, вызванное дефицитом гомогентизат-1,2-диоксигеназы*. При этом образуются большие количества гомогентизиновой кислоты (см. рис. 1), которая выделяется с мочой/накапливается в тканях.
P.S. * Ген гомогентизат-1,2-диоксигеназы расположен в хромосоме 3q13.3. Алкаптонурия чаще всего встречается в Доминиканской Республике и Словакии.
Основными клиническими проявлениями алкаптонурии у взрослых являются охроноз и артрит. Единственный симптом у детей — потемнение мочи при отстаивании, обусловленное окислением и полимеризацией гомогентизиновой кислоты. Данное заболевание можно заподозрить при наличии серых/черных пятен в подгузниках. Такой симптом может остаться незамеченным, поэтому диагноз часто остается не выявленным вплоть до взрослого возраста.
В результате накопления черного полимера гомогентизиновой кислоты возникает охроноз, который клинически проявляется в виде темных пятен на склере/ ушной раковине. Еще одним результатом такого накопления является артрит, который с возрастом может стать причиной инвалидности. Артрит поражает позвоночник и крупные суставы (плечевые, бедренные и коленные), при этом у мужчин он обычно проявляется в более тяжелой форме. Как и РА, алкаптонурический артрит имеет периоды обострения, а также рентгенологические данные, типичные для остеоартрита с характерным сужением суставных щелей и кальцификацией межпозвонковых дисков. Сообщалось о высокой частоте ССЗ (митральный и аортный вальвулит, кальцификация сердечных клапанов, ИМ).
Диагноз подтверждается значительной экскрецией гомогентизиновой кислоты в анализе мочи на содержание органических кислот. Содержание тирозина остается в норме. Фермент экспрессируется только в печени и почках.
Лечение артрита симптоматическое. При алкаптонурии нитизинон эффективно снижает синтез гомогентизиновой кислоты. При выявлении доклинических состояний целесообразно лечение нитизиноном и соблюдение диеты с ограничением ФА и тирозина, хотя долгосрочная эффективность не подтверждена.
ж) Дефицит тирозингидроксилазы. См. отдельную статью на сайте - просим вас пользоваться формой поиска по сайту выше.
з) Альбинизм. Альбинизм обусловлен дефицитом меланина, который является основным пигментом кожи и глаз (табл. 1). Меланин синтезируется меланоцитами из тирозина в клеточной органелле, связанной с мембраной (меланосоме). Меланоциты закладываются в эмбриональном нервном гребне и мигрируют в кожу, глаза (сосудистую и радужную оболочки), волосяные фолликулы и внутреннее ухо. Расположение меланина в глазах ограничено стромой радужной оболочки и пигментным эпителием сетчатки, а в коже и волосяных фолликулах он секретируется в эпидермис и стержни волос. Причиной альбинизма м.б. дефицит синтеза меланина, некоторые наследственные дефекты меланосом и нарушения миграции меланоцитов. Биосинтез меланина и многие аспекты клеточной биологии меланоцитов до конца не изучены (см. рис. 2). Конечными продуктами являются два пигмента: феомеланин (желто-красный пигмент) и эумеланин (коричнево-черный пигмент).
Клинически первичный альбинизм м.б. генерализо-ванным/локализованным. Первичный генерализованный альбинизм м.б. глазным/КГА. Альбинизм проявляется при некоторых синдромах в сочетании с тромбоцитарными, иммунологическими и неврологическими нарушениями. Для людей с генерализованным альбинизмом не характерна ни общая (загар), ни локальная (пигментные невусы) пигментация кожи.
Диагноз альбинизма обычно очевиден, но среди некоторых белокожих детей, чьим семьям присуща особенно светлая кожа, проводят ДД с альбинизмом. В отличие от пациентов с альбинизмом, у обычных детей со светлой кожей с возрастом прогрессивно развивается пигментация, у них нет глазных проявлений альбинизма, а пигментация аналогична таковой у др. членов семьи. Для клинической диагностики КГА, в отличие от др. типов кожной гипопигментации, необходимо наличие характерных глазных симптомов.
К глазным проявлениям альбинизма относятся: гипопигментация радужной оболочки и сетчатки с гипоплазией макулы, снижение остроты зрения, аномалии рефракции, нистагм, перемежающееся косоглазие и трансиллюминация радужной оболочки (диффузный красноватый оттенок радужной оболочки при офтальмоскопическом исследовании/при осмотре с помощью щелевой лампы). Также наблюдается аномальное прохождение оптических волокон в хиазме. В отличие от людей с нормальной пигментацией, у пациентов с альбинизмом большинство нервных волокон височной стороны сетчатки пересекаются с противоположным полушарием ГМ. Это приводит к отсутствию бинокулярного (стереоскопического) зрения и восприятия глубины, а также к многократному переключению зрения с глаза на глаз, вызывая перемежающееся косоглазие. Эта аномалия приводит к характерной картине зрительных вызванных потенциалов. Эти признаки являются специфичными для альбинизма и используются для диагностики.
Пациентам с КГА рекомендуется регулярное офтальмологическое наблюдение. Коррекция аномалий рефракции позволяет улучшить зрительную функцию. Обычно перемежающееся косоглазие не вызывает амблиопию и не требует хирургического вмешательства.
Пациентам с альбинизмом рекомендуется избегать УФО путем использования защитной одежды с длинными рукавами и солнцезащитных кремов с фотозащитным фактором (SPF; англ. Sun Protection Factor) >30. Меланин также присутствует в улитке внутреннего уха. Люди с альбинизмом м.б. более восприимчивыми к ототоксическим ЛП, таким как гентамицин.
КГА наследуется по АуР-типу. Выявлено множество клинических форм. Некоторые, кажущиеся различными, вызваны вариабельностью одного и того же гена. В меланогенез вовлечены несколько генов, расположенных в разных хромосомах (см. табл. 1). Попытки дифференцировать альбинизм на основе типа наследования, активности тирозиназы и степени гипопигментации не привели к появлению единой классификации. Приведенная ниже классификация основана на степени распределения меланина в организме и генах, вовлеченных в патогенез.
Для большинства генов альбинизма существует доступный в клинической практике генетический анализ (см. табл. 1). Молекулярная диагностика имеет небольшую терапевтическую значимость при изолированном альбинизме, но м.б. полезна для генетического консультирования семей.
1. Кожно-глазной (генерализованный) альбинизм. Наблюдается общий дефицит пигмента, заболевание поражает кожу, волосы и глаза. Идентифицировано не <4 генетически разл. форм кожно-глазного альбинизма: КГА1, КГА2, КГАЗ и КГА4. Пигмент полностью отсутствует у пациентов с КГА1А; др. типы м.б. неотличимы друг от друга клинически. У всех пациентов с данной патологией наблюдаются глазные симптомы. Все формы наследуются по АуР-типу.
- Кожно-глазной альбинизм типа 1 (тирозиназодефицитный альбинизм). У пациентов с ГКА1 нарушение обусловлено геном тирозиназы (TYR), расположенным в хромосоме 11q14.3. Было выявлено множество мутантных аллелей. Большинство пациентов с таким нарушением являются сложными гетерозиготами. Клиническим ДК КГА1 считается полное отсутствие пигмента при рождении. Состояние можно подразделить на КГА1А и КГА1В, в зависимости от активности фермента и возрастных различий в клинических проявлениях.
- Кожно-глазной альбинизм типа 1А (тирозиназонегативный кожно-глазной альбинизм). У пациентов с КГА1А, наиболее тяжелой формой КГА, оба аллеля TYR имеют патогенные варианты, полностью ингибирующие тирозиназу. Клинически наблюдается отсутствие пигмента в коже (молочно-белый цвет), волосах (белые волосы) и глазах (красно-серые радужки), проявляется при рождении и остается неизменным на протяжении всей жизни. Такие пациенты не загорают, у них не появляются пигментные невусы/веснушки.
- Кожно-глазной альбинизм типа 1В. Пациенты с КГА1В имеют патогенные варианты гена TYR, сохраняющие некоторую остаточную активность. Клинически у них полностью отсутствует пигмент при рождении, но с возрастом они становятся светло-русыми с голубыми/карими глазами. У них появляются пигментные невусы и веснушки, они могут загореть. Пациентов с КГА1В, в зависимости от степени пигментации, раньше разделяли на несколько подгрупп и считали генетически различными.
- Кожно-глазной альбинизм типа 2 (тирозиназопозитивный кожно-глазной альбинизм). КГА2 представляет собой наиболее распространенную форму генерализованного КГА, особенно у пациентов африканского происхождения. Клинически фенотип сильно варьирует; у большинства пациентов наблюдается некоторая пигментация кожи и глаз при рождении, они продолжают накапливать пигмент на протяжении всей жизни. У таких пациентов при рождении желтоватые волосы, которые с возрастом могут потемнеть. Они также могут иметь пигментные невусы и веснушки, а некоторые могут загорать. Клинически это состояние м.б. неотличимо от КГА1В. Однако пациенты с КГА2 имеют нормальную активность тирозиназы в волосяных луковицах. Дефект находится в гене ОСА2, который является ортологом гена р (снижение цвета красных глаз) у мышей. Этот ген синтезирует белок Р (белок мембраны меланосомы). Пациенты с синдромами Прадера-Вилли (Prader-Willi) и Ангельмана (Angelman), вызван ными микроделецией хромосомы 15q12, которая включает ген ОСА2, имеют небольшой дефицит пигмента.
- Кожно-глазной альбинизм типа 3 (рыжий альбинизм). Такая форма выявлена преимущественно у африканцев, афроамериканцев и уроженцев Новой Гвинеи. Пациенты с КГА3 могут вырабатывать феомеланин, но не эумеланин. Для взрослых пациентов характерны рыжеватые волосы и рыжевато-коричневая кожа. Цвет кожи специфичен для данной формы. Цвет кожи молодых людей может напоминать цвет кожи при КГА2. Патогенный вариант находится в гене тирозиназа-связывающего протеина 1 (TYRP1) (расположенного в хромосоме 9р23), функция которого до конца не изучена.
- Кожно-глазной альбинизм типа 4. КГА2-подобные проявления (как на коже, так и в глазах) наблюдались у пациентов с КГА4 (в основном из Японии) с дефектом в гене SLC45A2 (прежнее название МАТР) (расположенном в хромосоме 5р13.2).
2. Глазной альбинизм. У пациентов с глазным альбинизмом в первые месяцы жизни наблюдается нистагм, гипопигментация радужной оболочки и глазного дна, гипоплазия макулы, снижение остроты зрения. При исследовании биоптатов кожи/образцов корней волос методом электронной микроскопии наблюдаются характерные макромеланосомы. Большинство пациентов, страдающих глазным альбинизмом, имеют глазной альбинизм 1-го типа, представляющий собой заболевание, сцепленное с Х-хромосомой и вызванное патогенными вариантами в гене GPR143. Также существует редкая форма глазного альбинизма с поздним началом нейросенсорной глухоты и АуД-типом наследования.
- Глазной альбинизм типа 1 (глазной альбинизм Неттелшопа-Фоллса). Глазной альбинизм типа 1 представляет собой заболевание, сцепленное с Х-хромосомой и характеризующееся врожденным нистагмом, снижением пигментации структур глаза и нарушением зрения у пациентов мужского пола. Гетерозиготные женщины могут иметь участки аномальной пигментации сетчатки. В некоторых случаях, в зависимости от характера инактивации Х-хромосомы, у гетерозиготных женщин могут также наблюдаться тяжелые проявления, в т.ч. нистагм, гипопигментация радужной оболочки и макулы, гипоплазия макулы и снижение остроты зрения. В семьях с более темным цветом кожи может наблюдаться легкая степень гипопигментации кожи. Заподозрить глазной альбинизм типа 1 можно, если у пациентов мужского пола наблюдаются признаки глазного альбинизма, нормальная/умеренно сниженная пигментация кожи, а также семейный анамнез заболевания, свидетельствующий об Х-сцепленном наследовании. Это не прогрессирующее заболевание, с возрастом состояние глаз часто улучшается.
Если патология появляется у пациента впервые в его семье, то для подтверждения диагноза проводят исследование для выявления гена GPR143 (Хр22.2).
3. Синдромальные формы генерализованного альбинизма:
- Синдром Германски-Пудлака. Эта группа АуР-заболеваний вызывается патогенными вариантами одного из девяти разл. генов, расположенных в разных хромосомах — от HPS1* до HPS9. Синдром Германски-Пудлака можно заподозрить у пациентов с альбинизмом и геморрагическим диатезом и ВЗК/фиброзом легких. Подтип заболевания устанавливают при проведении молекулярных исследований.
Гены HPS необходимы для нормальной структуры и функционирования органелл, полученных из лизосом, в т.ч. меланосом и плотных тел тромбоцитов. У пациентов наблюдается тирозиназа-«+» КГА разл. степени тяжести, сопровождающийся дисфункцией тромбоцитов (вызванной отсутствием плотных тел тромбоцитов). В тканях накапливается цероидоподобное в-во. Синдром Германски-Пудлака не зависит от этнической принадлежности. Тем не менее учет происхождения пациентов может помочь в разработке экономически эффективной стратегии скрининга. Синдром Германски-Пудлака чаще всего встречается в двух регионах Пуэрто-Рико (1-й тип — в северных и 3-й тип — в центральных регионах в результате частных случаев генетического дрейфа). Наблюдаются кожные и глазные симптомы альбинизма. У пациентов могут развиться носовое и послеоперационное кровотечение и обильные менструации. Время свертывания крови увеличено, но число тромбоцитов в норме. К основным осложнениям относятся прогрессирующий фиброз легких у молодых людей и ВЗК-подобные заболевания по типу болезни Крона (Crohn) у подростков и молодых людей. Кроме того, отмечается почечная недостаточность и кардиомиопатия. При синдроме Германски-Пудлака 2-го типа наблюдается нейтропения. Лечение симптоматическое.
- Синдром Чедиака-Хигаши. Пациенты с этим редким АуР-заболеванием страдают КГА разл. степени тяжести и восприимчивы к инфекции. Часто наблюдаются бактериальные инфекции кожи и ВДП. В мазке крови в гранулоцитах можно увидеть гигантские пероксидаза-«+» лизосомальные гранулы. У пациентов снижено число меланосом аномально больших размеров (макромеланосомы). Склонность к кровотечению обычно умеренная. При отсутствии ответа на лечение у детей развивается стадия заболевания, известная как фаза акселерации, которая является серьезным жизнеугрожающим осложнением синдрома Чедиака-Хигаши. Данное состояние развивается вследствие активации макрофагов, приводящей к гемофагоцитарному лимфогистиоцитозу, а системные проявления включают лихорадку, лимфаденопатию, гепатоспленомегалию, цитопению и повышение уровня ферритина в плазме. У пациентов, выживших в детстве, могут развиться атрофия мозжечка, периферическая невропатия и задержка психического развития. Единственной известной причиной этого синдрома являются дефект гена LYST в хромосоме 1q42.3. Трансплантация стволовых кроветворных клеток м.б. эффективным методом лечения, позволяющим контролировать иммунодефицит и гематологические нарушения, а также предотвращать развитие фазы акселерации.
- Другие проявления генерализованного альбинизма. Гипопигментация является признаком др. синдромов, некоторые из них обусловлены нарушениями лизосомального биосинтеза/биологии меланосом. Пациентов с синдромом Грисцелли (Griscelli) отличают серебристо-серые волосы, сниженное содержание пигмента в коже и скопление меланосом в стержнях волос и центре меланоцитов, умственная отсталость и активация макрофагов с гемофагоцитозом разл. подтипов. У пациентов с синдромом Вичи (Vici) сочетаются иммунодефицит, умственная отсталость, агенезия мозолистого тела, катаракта и расщепление губы и неба. Для пациентов с дефицитом белка, взаимодействующего с МАРВР, характерен низкий рост, рецидивирующие инфекции, нейтропения.
4. Локализованный альбинизм. Локализованный альбинизм проявляется локальными участками гипопигментации кожи и волос, которые могут проявляться при рождении/развиваться со временем. Причиной этого состояния является аномальная миграция меланоцитов в период эмбрионального развития.
- Пиебалдизм. Пиебалдизм представляет собой АуД-наследственное заболевание, при котором ребенок обычно рождается с белой прядью волос на лбу. Кожа депигментирована и не содержит меланоцитов. Кроме того, обычно наблюдаются белые пятна на лице, туловище и конечностях. У пациентов с таким заболеванием были выявлены патогенные варианты генов KIT и SNAI2.
- Синдром Ваарденбурга. При синдроме Ваарденбурга белая прядь волос часто сочетается с боковым смещением внутренних уголков глаз, широкой переносицей, гетерохромией радужной оболочки и нейросенсорной тугоухостью. Это заболевание наследуется по АуД-типу; различают четыре основных типа. Пациенты с синдромом Ваарденбурга типа 1 (распространенная форма) имеют все предыдущие клинические симптомы, в т.ч. боковое смещение внутренних уголков глаз. Причиной заболевания являются патогенные варианты (>90%) гена РАХЗ. Пациенты с синдромом Ваарденбурга типа 2 имеют клинические признаки типа 1, кроме бокового смещения внутренних уголков глаз. Генетически это гетерогенное состояние, вызванное патогенными вариантами в нескольких генах, в т.ч. MITF, SOX10 и SNAI2. У пациентов с синдромом Ваарденбурга типа 3 наблюдаются все симптомы, присущие пациентам с типом 1, а также гипоплазия и контрактуры верхних конечностей. Причиной синдрома этого типа являются гетерозиготные/гомозиготные патогенные варианты гена РАХЗ. Синдром Ваарденбурга типа 4, сочетающийся с болезнью Гиршпрунга (Hirschsprung), генетически неоднороден; у разных пациентов были выявлены дефекты в разных генах (EDN3, EDNRB и SOX 10).
К др. причинам локальной гипопигментации относятся витилиго и гипомеланоз Ито (Ito).