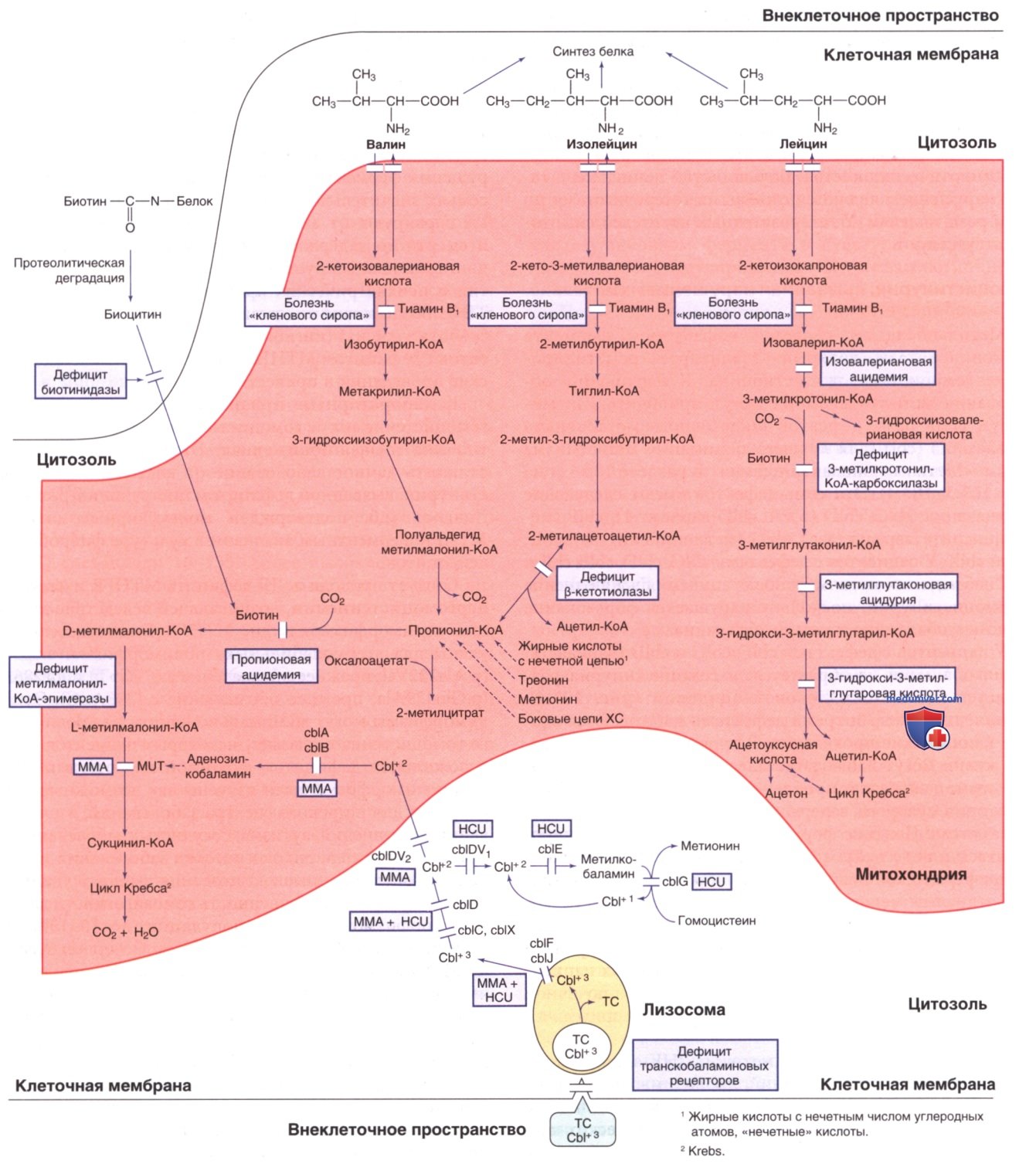
Начальные стадии распада аминокислот с разветвленной цепью (АКРЦ) — изолейцина, лейцина и валина — являются аналогичными (см. рис. 4). АКРЦ в мышечной ткани в катаболических условиях подвергаются обратимой реакции трансаминирования, катализируемой трансаминазой АКРЦ. α-Кетокислоты, образованные в результате данной реакции, затем подвергаются стадии окислительного декарбоксилирования, опосредованной комплексом дегидрогеназы α-кетокислоты с разветвленной цепью (ДАКРЦ). Дефицит ДАКРЦ приводит к развитию болезни «кленового сиропа», тогда как дефицит ферментов, опосредующих более отдаленные стадии, приводит к накоплению фермент-специфических органических кислот, выделяемых с мочой, т.о., давая этим врожденным ошибкам метаболизма эпонимы: органические ацидемии и органические ацидурии.
Рисунок 4. Пути метаболизма аминокислот с разветвленной цепью, биотина и витамина В12 (кобаламина). Cbl — кобаламин; cbl — нарушение метаболизма кобаламина; cblDV1 — кобаламин D (вариант 1); cblDV2 — кобаламин D (вариант 2); HCU — гомоцистинурия; ММА — метилмалоновая ацидемия; MUT — мутаза; OHcbl — гидроксокобаламин; ТС — транскобаламин;TCR — рецептор транскобаламина. Названия ферментов приведены в тексте
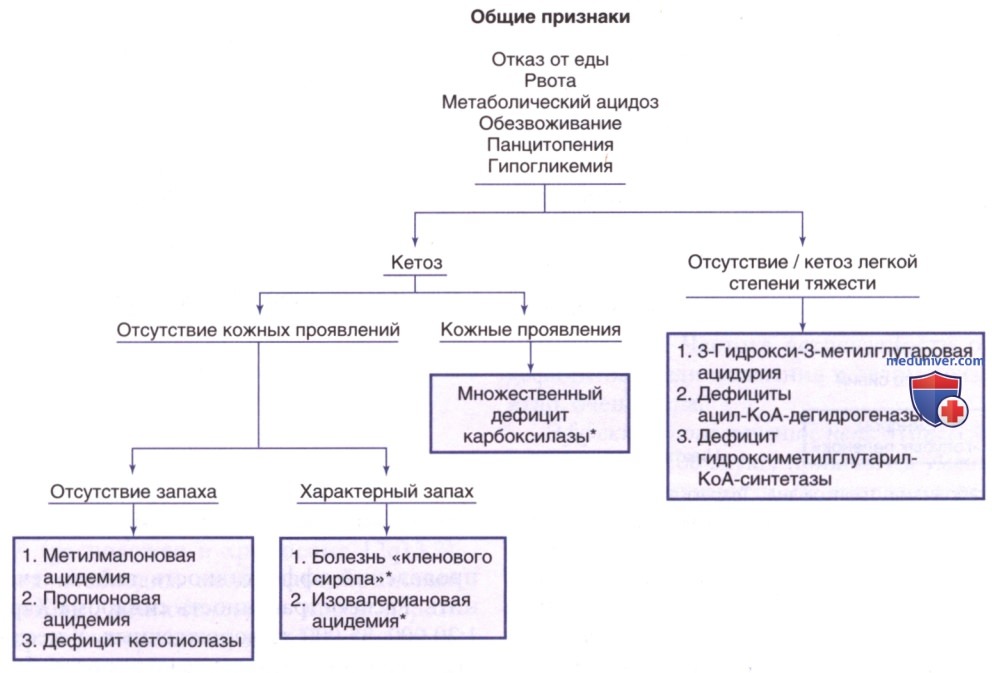
Данные нарушения обычно приводят к метаболическому ацидозу, возникающему в первые несколько дней жизни. Хотя большинство клинических проявлений неспецифичны, некоторые симптомы позволяют объяснить природу дефицита фермента. На рис. 6 представлен подход к диагностике у младенцев с подозрением на органическую ацидемию. Диагностика основана на выявлении и измерении специфических органических кислот в жидкостях организма (кровь, моча), обнаружении мутаций в соответствующем гене и измерении активности ферментов.
Рисунок 6. Клиническая картина и диагностика органической ацидемии у новорожденных. Звездочками отмечены нарушения, при которых у пациентов появляется характерный запах (см. текст и табл. 2)
Органические ацидемии не ограничиваются дефектами катаболических путей АКРЦ. Нарушения, вызывающие накопление др. органических кислот, включают в т.ч. нарушения метаболизма производных лизина, γ-глутаминового цикла, заболевания, вызванные нарушениями метаболизма молочной кислоты, и дикарбоновые ацидемии, обусловленные дефектами распада жирных кислот.
а) Болезнь "Кленового сиропа". Декарбоксилирование лейцина, изолейцина и валина осуществляется сложным комплексом ферментной системы ДАКРЦ, использующей тиамина пирофосфат (витамин B1) в качестве кофермента. Данный митохондриальный фермент состоит из четырех субъединиц: E1a, E1β, Е2 и ЕЗ. Субъединица Е3 является общей с двумя др. дегидрогеназами — пируватдегидрогеназой и а-кетоглутаратдегидрогеназой. Дефицит любой из этих субъединиц вызывает болезнь «кленового сиропа» (см. рис. 4) — заболевание, названное из-за сладкого запаха кленового сиропа, присущего всем жидкостям организма, особенно моче, при данной патологии. Заболевания, вызванные дефектами E1β, E1β, Е2 и Е3, обозначаются как лейциноз IА-типа, IB-типа, 2-го типа и 3-го типа соответственно.
Однако с клинической точки зрения эта классификация не очень информативна, поскольку тяжесть клинических проявлений не коррелирует с дефектом какой-либо отдельной субъединицы фермента и не соответствует ей. У младенца с дефектом IА-типа клинические проявления м.б. от относительно легкого до очень тяжелого. Более информативная классификация основана на клинических данных и ответе на терапию тиамином и выделяет пять фенотипов данного заболевания.
1. Классическая болезнь «кленового сиропа». Классическая болезнь «кленового сиропа» имеет наиболее тяжелую клиническую манифестацию. Активность комплекса ДАКРЦ в данной группе составляет 0-2% в сравнении с контрольной группой. У пациентов с не-контролируемым/трудноконтролируемым течением заболевания развиваются признаки острой энцефалопатии. В основе данного жизнеугрожающего осложнения лежат сложные механизмы, но ключевыми факторами острой энцефалопатии, по всей видимости, являются лейцин и его производное (α-кетоизокапроновая кислота).
Повышенный уровень лейцина конкурентно ингибирует захват др. аминокислот транспортным белком больших нейтральных аминокислот. После поступления в ткань ГМ лейцин метаболизируется аминотрансферазой АКРЦ до α-кетоизокапроновой кислоты, что приводит к нарушению метаболизма нейромедиаторов и аминокислот (глутамата, ГАМК, глутамина, аланина и аспартата). а-Кетоизокапроновая кислота может обратимо ингибировать процесс окислительного фосфорилирования, что приводит к церебральному лактат-ацидозу. Описанные процессы в совокупности нарушают нормальное функционирование нейронов и нейроглии, что клинически проявляется в виде энцефалопатии и ОГМ и носит название лейциноз. Младенцы с данной патологией выглядят здоровыми при рождении, но в первые дни жизни у них развиваются проблемы с кормлением и рвота.
В течение нескольких дней ребенок может стать вялым и впасть в кому. При физикальном осмотре наблюдаются гипертонус и ригидность мышц с тяжелым опистотонусом. Периоды гипертонуса могут чередоваться с приступами вялости, проявляющейся в виде повторяющихся движений конечностей («боксирование» и «езда на велосипеде»). Неврологические симптомы часто ошибочно принимают за генерализованный сепсис и менингит. Также может наблюдаться ОГМ; у большинства младенцев возникают судороги, часто встречается гипогликемия. В отличие от большинства гипогликемий, коррекция уровня глюкозы в крови не улучшает клиническое состояние. Результаты общеклинического исследования обычно в норме, за исключением уровня глюкозы в крови и кетоацидоза разл. степени выраженности. При отсутствии лечения смерть может наступить в первые несколько не-дель/месяцев жизни.
Часто заподозрить данную патологию можно при обнаружении специфического запаха кленового сиропа у пота и ушной серы. Для подтверждения диагноза обычно проводят исследование аминокислот в плазме: заметно повышено содержание лейцина, изолейцина, валина и аллоизолейцина (стереоизомера изолейцина, обычно не обнаруживаемого в крови) и сниженное содержание аланина. Концентрация лейцина обычно выше, чем содержание трех др. аминокислот. В моче повышено содержание лейцина, изолейцина и валина и их соответствующих кетокислот. Указанные кетокислоты можно определить качественно при добавлении в мочу нескольких капель реагента 2,4-динитрофенилгидразина (0,1% в 0,1 н. HCl). В случае «+» реакции образуется желтый осадок 2,4-динитрофенилгидразона.
В остром периоде ОГМ м.б. обнаружен методами визуализации, наиболее выражен отек в мозжечке, на дорсальной поверхности ствола ГМ, ножке ГМ и внутренней капсуле. В восстановительный период, а также с возрастом при визуализации ГМ наблюдаются гипомиелинизация и церебральная атрофия.
Лечение в остром периоде направлено на восполнение потери жидкости и быструю элиминацию АКРЦ и их метаболитов из тканей и жидкостей организма. Ключевым метаболическим звеном патогенеза энцефалопатии при болезни «кленового сиропа», по всей видимости, является накопление тканью ГМ лейцина и последующего метаболита (а-кетоизокапроновой кислоты). Следовательно, стратегии лечения для контроля острых и хронических проявлений заболевания направлены на снижение уровня лейцина в плазме.
Восполнение потери жидкости само по себе не дает хорошего клинического эффекта из-за низкого почечного клиренса лейцина. Наиболее эффективным методом лечения тяжелобольных младенцев является гемодиализ, который следует назначать незамедлительно; значительное снижение уровней лейцина, изолейцина и валина в плазме обычно наблюдается в течение 24 ч. Как можно раньше следует начать в/в введение/прием внутрь достаточного количества калорий и питательных в-в с целью купирования катаболического статуса пациента. При ОГМ может потребоваться лечение маннитолом, диуретиками (напр., фуросемидом) и гипертоническим р-ром. Как ни странно, добавление изолейцина и валина необходимо для контроля уровня лейцина в плазме пациентов с данной патологией. Изолейцин и валин, введенные в подобранных дозах, конкурируют с лейцином за транспортный белок больших нейтральных аминокислот на ГЭБ и, т.о., уменьшают проникновение лейцина в ЦНС и помогают в профилактике и лечении лейциновой энцефалопатии.
После выхода из острой стадии требуется соблюдение диеты с низким содержанием АКРЦ. В продаже имеются синтетические формулы, не содержащие лейцин, изолейцин и валин. Поскольку эти аминокислоты не синтезируются эндогенно, необходимо обеспечить АКРЦ в рационе в виде полноценного белка в количестве, соответствующем возрасту пациента. Для предотвращения дефицита незаменимых аминокислот необходимо тщательно титровать их количество, проводя определение их уровней в плазме и уделяя особое внимание содержанию изолейцина, лейцина и валина. У младенцев с очень низким содержанием изолейцина/валина в плазме возникает клиническое состояние, напоминающее энтеропатический акродерматит. Добавление в диету изолейцина/валина соответственно способствует исчезновению кожной сыпи.
Пациентам с болезнью кленового сиропа необходимо соблюдать диету на протяжении всей жизни. Пациентам с классической формой заболевания проводилась трансплантация печени, получены обнадеживающие результаты.
Долгосрочный прогноз у детей с данной патологией остается неоднозначным. Любая стрессовая ситуация, напр. инфекция/операция, особенно в 6-12 лет, может привести к тяжелому кетоацидозу, ОГМ и летальному исходу. Частыми последствиями являются когнитивные и др. неврологические нарушения.
2. Промежуточная (легкая) форма болезни «кленового сиропа». У детей с промежуточной формой болезни «кленового сиропа» заболевание развивается после неонатального периода и протекает легче. Клинические проявления манифестируют постепенно и ограничиваются ЦНС. У пациентов присутствует умственная отсталость от легкой до умеренной степени, с/без судорог. Появляется запах кленового сиропа, а в моче обнаруживается умеренное количество АКРЦ и их производных кетокислот. Концентрации лейцина, изолейцина и валина в плазме умеренно повышены, а концентрации лактата и пирувата обычно остаются нормальными. Данную патологию часто диагностируют у детей при лечении сопутствующего заболевания, при котором могут проявиться признаки и симптомы классической болезни «кленового сиропа». Активность дегидрогеназы составляет 3-40% от нормы.
Поскольку у пациентов с тиамин-чувствительной формой симптоматика схожа с промежуточной (легкой) формой, рекомендуется попробовать терапию тиамином. Показана такая же диетотерапия, как при классической болезни «кленового сиропа».
3. Интермиттирующая форма болезни «кленового сиропа». При интермиттирующей форме у кажущихся здоровыми детей при любом стрессовом и катаболическом состоянии, напр. инфекции/операции, появляются рвота, запах кленового сиропа, атаксия, летаргия, кома.
Также возможно при высокобелковой диете, интеркуррентном заболевании*.
P.S. * Федеральные КР по диагностике и лечению болезни кленового сиропа. П.В. Новиков, Е.А. Николаева, Т.Э. Боровик, Т.В. Бушуева, Е.Ю. Захарова. 2013 г. С. 25 (КР 2016 и 2019 гг. не утверждены).
Во время таких приступов лабораторные показатели те же, что и при классической форме, данное состояние может привести к летальному исходу. Лечение острого приступа интермиттирующей формы болезни аналогично лечению классической формы. После выхода из острой стадии рекомендуется диета с низким содержанием АКРЦ, даже если пациент хорошо переносит обычное питание. Активность ДАКРЦ у пациентов при этой форме выше, чем при классической, и может достигать 40% от нормы.
4. Тиамин-чувствительная болезнь «кленового сиропа». У некоторых детей с легкой/средней формой болезни «кленового сиропа», получающих высокие дозы тиамина, наблюдается резкое улучшение клинических и биохим. показателей. Хотя некоторые пациенты поддаются лечению тиамином 10 мг/сут, другим для достижения «+» результата может потребоваться до 100 мг/сут как минимум 3 нед. Пациентам с данной патологией необходима диета с низким содержанием АКРЦ. Ферментативная активность у таких пациентов не >40% от нормы.
5. Болезнь «кленового сиропа», обусловленная дефицитом субъединицы Е3-протеина и сопровождающаяся лактат-ацидозом (болезнь «кленового сиропа» 3-го типа). Несмотря на название «болезнь “кленового сиропа” 3-го типа», это очень редкое нарушение приводит к клиническим проявлениям и биохим. сдвигам, охватывающим широкий спектр митохондриальных реакций. Субъединица Е3 (дигидролипоамиддегидрогеназа) является компонентом комплекса ДАКРЦ, комплекса пируватдегидрогеназы и комплекса а-кетоглутаратдегидрогеназы. Дефекты дигидролипоамиддегидрогеназы приводят к лактат-ацидозу, повышению уровня пирувата, а также признакам и симптомам, похожим на промежуточную форму болезни «кленового сиропа». В 2 мес прогрессируют неврологические нарушения, проявляющиеся гипотонией и задержкой развития. Прогрессирование аномальных движений приводит к развитию атаксии/синдрому Ли (Leigh). Смерть может наступить в раннем детстве.
К лабораторным признакам относится стойкий лактат-ацидоз с высоким уровнем пирувата и аланина в плазме. Концентрации АКРЦ в плазме умеренно повышены. В моче пациентов наблюдается повышенная экскреция лактата, пирувата, α-кетоглутарата и трех кетокислот с разветвленной цепью.
Эффективного лечения нет. Диеты с низким содержанием АКРЦ и терапия высокими дозами тиамина, биотина и липоевой кислоты оказались неэффективными.
6. Генетика и распространенность болезни «кленового сиропа». Все формы данной патологии наследуются по АуР-типу. Гены разных субъединиц находятся в разных хромосомах. Ген E1α (BCKDHA) расположен в хромосоме 19ql3.2; ген Е1β (BCKDHB) находится в хромосоме 6q14.1; ген для Е2 (DBT) расположен в хромосоме 1р21.2; ген Е3 (DLD) находится в хромосоме 7q31.1. Корреляции «генотип-фенотип» трудно установить, обычно они неточны. Исключением является тиамин-чувствительная форма болезни, которая, как было показано, обусловлена дефектом гена DBT. У большинства пациентов наблюдаются сложные гетерозиготные состояния, наследуемые по двум разл. патогенным аллелям. В -80% случаев наблюдаются мутации BCKDHA (45%) и BCKDHB (35%). В 20% случаев причиной являются мутации DBT.
Распространенность, по оценкам, составляет 1:185 000 живорожденных. Классическая форма в США распространена у меннонитов старого порядка и встречается в 1:380 живорожденных. В данной популяции заболевшие пациенты гомозиготны по конкретной мутации (с.1312Т>А) в субъединице Е1а, кодирующей BCKDHA.
Болезнь «кленового сиропа» можно выявить на ранней стадии по результатам скрининга новорожденных. Однако в большинстве случаев, особенно при классической форме, заболевание может уже манифестировать к моменту получения результатов скрининга. Пренатальную диагностику проводят с помощью ферментативного анализа культуры амниоцитов, культуры тканей хориальных ворсинок и непосредственного анализа образцов хориальных ворсинок для определения известных мутаций пораженного гена.
Известно о нескольких успешных случаях беременности у женщин с разл. формами данного заболевания. Тератогенный потенциал лейцина при беременности неизвестен. Важным для минимизации риска метаболической декомпенсации и оптимизации питания плода является строгий контроль изолейцина, лейцина и валина до и вовремя беременности. Матери с болезнью «кленового сиропа» нуждаются в тщательном наблюдении и диетотерапии, коррекции электролитов и жидкости в послеродовой период.
б) Дефицит киназы α-кетоациддегидрогеназы с разветвленной цепью. Нарушение регуляции дегидрогеназы а-кетокислоты с разветвленной цепью (ДАКРЦ) с помощью ДАКРЦ-киназы (BCKDK), фермента, ответственного за инактивацию ДАКРЦ-комплекса, опосредованную фосфорилированием, вызывает обратный биохим. фенотип болезни «кленового сиропа». Дефекты BCKDK снижают «-» регуляцию киназой, что приводит к неконтролируемому расщеплению и истощению изолейцина, лейцина и валина в плазме и ГМ. У пациентов с дефицитом BCKDK наблюдаются низкие концентрации изолейцина, лейцина и валина в плазме, сопровождающиеся развитием РАС, интеллектуальных нарушений, проблемами с мелкой моторикой и судорогами.
в) Дефицит дегидрогеназы α-кетокислот с разветвленной цепью. Изолейцин, лейцин и валин транспортируются через ГЭБ в основном при помощи гетеродимерного транспортного белка больших нейтральных аминокислот LAT1, кодируемого SLC7A5. Дефект LAT1, вызванный мутацией SLC7A5, снижает концентрации изолейцина, лейцина и валина в ГМ. Пациенты с данным дефектом могут клинически напоминать пациентов с дефицитом BCKDK: манифестируют признаки РАС, микроцефалии, задержки развития крупной моторики и в некоторых случаях возникают судороги.
г) Изовалериановая ацидемия. Причиной изовалериановой ацидемии является дефицит изовалерил-КоА-дегидрогеназы (см. рис. 4). Снижение/потеря активности изовалерил-КоА-дегидрогеназы нарушает процесс распада лейцина. В жидкостях организма накапливаются производные изовалериановой кислоты, изовалерилкарнитина, изовалерилглицина и 3-гидроксиизовалериановой кислоты. Определение данных соединений позволяет проводить биохим. диагностику и скрининг. Клиническая манифестация изовалериановой ацидемии очень разнообразна, от практически бессимптомного до тяжелого течения. Внедрение скрининга новорожденных и проактивная (упреждающая) терапия изовалериановой ацидемии изменили исход и клиническое течение заболевания.
Выделяют две клинические формы заболевания — острую неонатальную (у 60-70% больных) и хроническую интермиттирующую*.
P.S. * КР «Изовалериановая ацидемия у детей», Союз педиатров России. 2018 г. С. 36.
В семьях новорожденных с манифестным заболеванием были описаны случаи, когда у старших братьев/сестер с идентичным генотипом и биохим. сдвигами отсутствовали клинические проявления. Это позволяет предположить, что выявление при скрининге новорожденных пациентов с этой патологией до появления симптомов может улучшить клинические исходы.
У пациентов с тяжелой формой изовалериановой ацидемии в младенческом возрасте могут развиваться рвота, тяжелый ацидоз, гипераммониемия, гипогликемия, гипокальциемия и угнетение функции костного мозга. При отсутствии надлежащего лечения могут развиться заторможенность, судороги и кома, что может приводить к летальному исходу. Рвота м.б. настолько сильной, что возникает подозрение на пилоростеноз. Может присутствовать характерный запах потных ног/прогорклого сыра. Младенцы, пережившие острую стадию заболевания, в старшем возрасте подвержены риску развития эпизодов метаболической декомпенсации. В легкой форме и при отсутствии лечения типичные клинические проявления тяжелой формы изовалериановой ацидемии (рвота, заторможенность, ацидоз и кома) могут отсутствовать до достижения ребенком нескольких месяцев/лет. Острые эпизоды метаболической декомпенсации могут возникать в период воздействия неблагоприятных факторов, которые обусловливают усиление процессов клеточного катаболизма, напр. при инфекции, обезвоживании, хирургическом вмешательстве, высокобелковой диете.
Также возможно при вакцинации, ФН/психоэмоциональной нагрузке и низкокалорийной диете.
Острые эпизоды заболевания м.б. ошибочно приняты за ДКА. У некоторых пациентов могут наблюдаться острые и рецидивирующие эпизоды панкреатита.
В период острых приступов лабораторными признаками являются кетоацидоз, нейтропения, тромбоцитопения и иногда панцитопения. У некоторых пациентов могут наблюдаться гипокальциемия, гипогликемия, уме-ренная/тяжелая степень гипераммониемии. Повышенный уровень аммиака в плазме может указывать на нарушение цикла мочевины. Однако при дефектах цикла мочевины у младенца обычно отсутствует значительный кетоацидоз (см. рис. 6).
Диагноз устанавливают на основании заметного повышения содержания метаболитов изовалериановой кислоты (изовалерилглицина, 3-гидроксиизовалериановой кислоты) в жидкостях организма, особенно в моче. Основным контролируемым соединением в плазме является изовалерилкарнитин (С5-карнитин). С5-карнитин можно измерить в сухих пятнах крови в рамках универсального скрининга новорожденных при использовании тандемной масс-спектрометрии. Молекулярный анализ гена IVD позволяет подтвердить диагноз. Некоторым пациентам с сомнительными результатами может потребоваться анализ активности фермента в культуре фибробластов кожи.
Лечение острого приступа направлено на восполнение потери жидкости, купирование катаболического состояния (путем обеспечения достаточного количества калорий внутрь/в/в), коррекцию метаболического ацидоза и облегчение экскреции изовалериановой кислоты. Прием L-карнитина (внутрь 100 мг/кг в сутки) повышает экскрецию изовалериановой кислоты за счет образования изовалерилкарнитина, который выводится с мочой. Поскольку изовалерилглицин имеет высокий клиренс с мочой, то для повышения его образования некоторые центры рекомендуют прием БАД глицина (250 мг/кг в сутки). В некоторых случаях м.б. эффективно временное ограничение потребления белка (<24 ч). Если у пациентов наблюдается значительная гипераммониемия (содержание аммиака в крови >200 мкмоль/л), то следует применять меры, снижающие содержание аммиака в крови. Если вышеуказанными мерами не удалось достичь значительного клинического и биохим. улучшения, то может потребоваться заместительная почечная терапия.
При долгосрочном лечении пациентов с изовалериановой ацидемией необходимо ограничить потребление белка в соответствии с возрастом (рекомендуемая диетическая норма белка). Положительно влияет прием БАД [карнитина ± глицина]. Своевременное и правильное лечение позволяет ребенку развиваться нормально.
Пренатальная диагностика м.б. выполнена методом ферментативного анализа в культуре амниоцитов/иссле-дованием гена IVD, если известны причинные мутации.
Проводится локализация гена IVD-15q13-q15.
Зарегистрированы случаи успешных беременностей с благоприятными исходами. В США и др. странах используется универсальный скрининг новорожденных на изовалериановую ацидемию. Данное заболевание вызывается АуР-патогенными вариантами гена IVD. По оценкам, частота встречаемости составляет от 1:62 500-250 000 живорождений (в некоторых частях Германии и в США соответственно).
д) Множественный дефицит карбоксилаз (дефекты цикла биотина). Биотин представляет собой водорастворимый витамин, являющийся кофактором всех четырех карбоксилаз у человека: пируваткарбоксилазы, ацетил-КоА-карбоксилазы, пропионил-КоА-карбоксилазы и 3-метилкротонил-КоА-карбоксилазы. Последние два фермента участвуют в метаболизме лейцина, изолейцина и валина (см. рис. 4).
Большая часть биотина, поступающего с пищей, содержится в белках. Свободный биотин вырабатывается в кишечнике под действием пищеварительных ферментов, кишечных бактерий и, вероятно, биотинидазы. Биотинидаза, которая содержится в сыворотке крови и большинстве тканей, важна для метаболизма биотина в организме, она высвобождает его из апоферментов (карбоксилаз; см. рис. 4). Свободный биотин должен образовывать ковалентную связь с апокарбоксилазами для образования активированного фермента (голокарбоксилазы). Это связывание катализируется синтетазой голокарбоксилаз. Недостаточная активность данного фермента/биотинидазы приводит к нарушению работы всех карбоксилаз и возникновению органических ацидемий.
1. Дефицит синтетазы голокарбоксилазы. У младенцев с этим редким АуР-заболеванием симптомы проявляются в первые несколько недель жизни. Симптомы могут появиться уже через несколько часов после рождения/позже, до 8 лет. Клинически у ребенка с данной патологией вскоре после рождения возникают расстройства дыхания (тахипноэ и апноэ). Также обычно наблюдаются проблемы с кормлением, рвота и гипотония. При отсутствии лечения могут возникнуть генерализованная эритематозная сыпь с шелушиванием и алопецией, задержка физического развития, повышенная возбудимость, судороги, заторможенность, вялость и даже кома. Часто наблюдается задержка психического развития. Иммунодефицит проявляется восприимчивостью к инфекциям. Моча может иметь специфический запах кошачьей мочи. Сыпь (при ее наличии) позволяет отличить это заболевание от др. органических ацидемий (см. рис. 6).
К лабораторным признакам относятся метаболический ацидоз, кетоз, гипераммониемия и присутствие разл. органических кислот (молочная кислота, 3-метилкротоновая кислота, 3-метилкротонилглицин, тиглилглицин, 3-ОН-пропионовая кислота, метилцитриновая кислота и 3-гидроксиизовалериановая кислота) в жидкостях организма. Диагноз подтверждается обнаружением мутации гена HLCS/определением активности фермента в лимфоцитах/культуре фибробластов. Большинство мутаций гена HLCS вызывают повышение значения Km (константа диссоциации Михаэлиса-Ментена (Michaelis-Menten)) фермента по отношению к биотину; ферментативную активность у таких пациентов можно восстановить при введении больших доз биотина.
Младенцев с дефицитом синтетазы голокарбоксилазы можно выявить во время скрининга новорожденных при обнаружении повышенного содержания С5-ОН-карнитина методом тандемной масс-спектрометрии. Ферментативный анализ биотинидазы у данных младенцев показывает нормальные результаты.
Лечение биотином (внутрь 10-20 мг/сут) обычно приводит к уменьшению клинической симптоматики и нормализации биохим. показателей. Ранняя диагностика и своевременное лечение являются критически важными факторами для предотвращения необратимых неврологических последствий. Однако у некоторых пациентов полного излечения достигнуть невозможно даже при применении больших доз биотина (до 60 мг/сут).
Ген синтетазы голокарбоксилазы (HLCS) расположен в хромосоме 21q22.13. Пренатальная диагностика м.б. выполнена методом пренатального молекулярного анализа известных мутаций HLCS/путем анализа активности ферментов в культуре амниотических клеток. Беременные матери, у старших детей которых был выявлен дефицит синтетазы голокарбоксилазы, на поздних сроках беременности получали лечение биотином. Младенцы рождались здоровыми, но эффективность дородового лечения остается неясной.
2. Дефицит биотинидазы. Нарушение активности биотинидазы приводит к дефициту биотина. У младенцев с данной патологией могут развиваться клинические проявления, похожие на дефицит синтетазы голокарбоксилазы. Однако, в отличие от последнего, симптомы дефицита биотинидазы обычно появляются позже, в возрасте нескольких месяцев/лет. Это происходит, вероятно, из-за наличия свободного биотина, полученного от матери/при кормлении. Клинические проявления в основном ограничиваются кожей и НС. Могут возникать атопический/себорейный дерматит, кандидоз, алопеция, атаксия, судороги (обычно миоклонические), гипотония, задержка психического развития, атрофия зрительного нерва, нейросенсорная тугоухость и иммунодефицит в результате нарушения функции Т-клеток. Зарегистрировано несколько случаев трудноизлечимого себорейного дерматита у детей с частичным (15-30%) дефицитом биотинидазы. Его удалось вылечить применением биотина; др. симптомов у них не наблюдалось.
Программы скрининга выявляли бессимптомных детей и взрослых с дефицитом данного фермента.
У большинства из них наблюдался частичный дефицит биотинидазы. Предполагается, что клинические проявления заболевания исчезают с внедрением универсального скрининга новорожденных, направленного на раннее выявление и лечение пациентов.
Лабораторные признаки и содержание органических кислот в жидкостях организма похожи на дефицит синтетазы голокарбоксилазы (см. выше). Диагноз устанавливается на основании измерения активности фермента в сыворотке крови/методом верификации мутантного гена. Лечение свободным биотином (5-20 мг/сут) позволяет достичь резкого клинического и биохим. улучшения. Пациентам с частичным дефицитом биотинидазы рекомендуется лечение биотином. По оценкам, частота встречаемости данного АуР-заболевания составляет 1:60 000 живорожденных. Ген биотинидазы (BTD) расположен в хромосоме 3р25.1. Пренатальная диагностика возможна на основании обнаружения мутации гена BTD/реже — методом измерения активности фермента в амниотических клетках, хотя на практике пренатальный подход используется редко.
3. Множественный дефицит карбоксилаз, вызванный приобретенным дефицитом биотина. Приобретенный дефицит биотина может развиваться у младенцев, получающих полное парентеральное питание без добавления биотина, у пациентов, длительно принимающих противоэпилептические ЛП (фенобарбитал, фенитоин, примидон, карбамазепин), а также у детей с синдромом короткой кишки/хронической диареей, получающих смеси с низким содержанием биотина. Дефицит биотина м.б. вызван чрезмерным употреблением сырых яиц, поскольку белок авидин в яичном белке связывает биотин, уменьшая его всасывание. У младенцев с дефицитом биотина может развиться дерматит, алопеция и кандидозные кожные инфекции. Это состояние легко поддается лечению приемом биотина внутрь.
е) Дефицит 3-метилкротонил-Коа-карбоксилазы. Указанный фермент является одной из четырех карбоксилаз, которым биотин необходим в качестве кофактора (см. рис. 4). Изолированный дефицит данного фермента необходимо отличать от нарушений метаболизма биотина (множественный дефицит карбоксилазы), которые вызывают снижение активности всех четырех карбоксилаз (см. ранее). 3-Метилкротонил-КоА-карбоксилаза представляет собой гетеромерный фермент, состоящий из α-(биотинсодержащих) и β-субъединиц, кодируемых генами МССС1 и МССС2 соответственно. У новорожденных дефицит 3-метилкротонил-КоА-карбоксилазы м.б. обнаружен путем выявления повышенного содержания С5-ОН в сухих пятнах крови. Универсальный скрининг новорожденных с использованием тандемной масс-спектрометрии выявил неожиданно большое количество младенцев с дефицитом 3-метилкротонил-КоА-карбоксилазы с частотой встречаемости 1:2400-68 000.
Клинические проявления сильно варьируют: от полностью бессимптомных взрослых (в т.ч. матерей заболевших новорожденных) до детей с задержкой психического развития без эпизодов метаболической декомпенсации и пациентов с судорогами, гипераммониемией и метаболическим ацидозом. При тяжелой форме дефицита 3-ме-тилкротонил-КоА-карбоксилазы у ребенка, казавшегося здоровым, после незначительной инфекции развивается острый эпизод рвоты, гипотонии, заторможенности и судорог, в некоторых случаях прогрессирующий до жизнеугрожающих осложнений (напр., синдрома Рея (Reye), комы). У пациентов, склонных к развитию данных симптомов, заболевание начинает проявляться в возрасте от 3 нед до 3 лет. Среди младенцев, выявленных при скрининге новорожденных, 85-90% детей остаются бессимптомными. Причина разл. исходов неизвестна. Ни один из описанных симптомов нельзя однозначно соотнести со степенью дефицита фермента.
К лабораторным признакам в период острых эпизодов относятся легкая/средняя степень метаболического ацидоза, кетоз, гипогликемия, гипераммониемия и повышенный уровень трансаминаз в сыворотке крови. В моче повышено содержание 3-гидроксиизовалериановой кислоты и 3-метилкротонилглицина. Экскреция 3-метилкротоновой кислоты с мочой при данном заболевании обычно в норме, поскольку накопленный 3-метилкротонил-КоА превращается в 3-гидроксиизовалериановую кислоту. Профиль ацилкарнитина в плазме показывает повышенное содержание С5-ОН. Часто встречается тяжелый вторичный дефицит карнитина. Дефицит 3-метилкротонилКоА-карбоксилазы следует биохимически отличать от множественного дефицита карбоксилаз, при котором, помимо 3-гидроксиизовалериановой кислоты, также присутствуют молочная кислота и метаболиты пропионовой кислоты.
Диагноз м.б. подтвержден молекулярным исследованием/измерением активности фермента в культуре фибробластов. Для исключения множественного дефицита карбоксилаз необходимо подтверждение нормальной активности др. карбоксилаз.
Лечение острых эпизодов сходно с лечением изовалериановой ацидемии (см. ранее). Меры принимаются незамедлительно и должны быть направлены на восполнение потери жидкости и коррекцию гипогликемии и тяжелого метаболического ацидоза путем инфузии декстрозы («Глюкозы») и натрия гидрокарбоната («Натрия бикарбоната»). Прием БАД L-карнитина позволяет устранить вторичный дефицит карнитина, наблюдаемый у 50% пациентов. Пациентам, имеющим клинические проявления, некоторые центры предлагают придерживаться рекомендуемой нормы потребления белка в сочетании с приемом L-карнитина внутрь и упреждающим контролем катаболических состояний. У большинства пациентов ожидается нормальный рост и развитие.
Дефицит 3-метилкротонил-КоА-карбоксилазы является АуР-заболеванием. Ген α-субъединицы (МССС1) расположен в хромосоме 3q27.1, а ген β-субъединицы (МССС2) находится в хромосоме 5q13.2. Мутации в любом из данных генов приводят к дефициту фермента со сходными клиническими признаками.
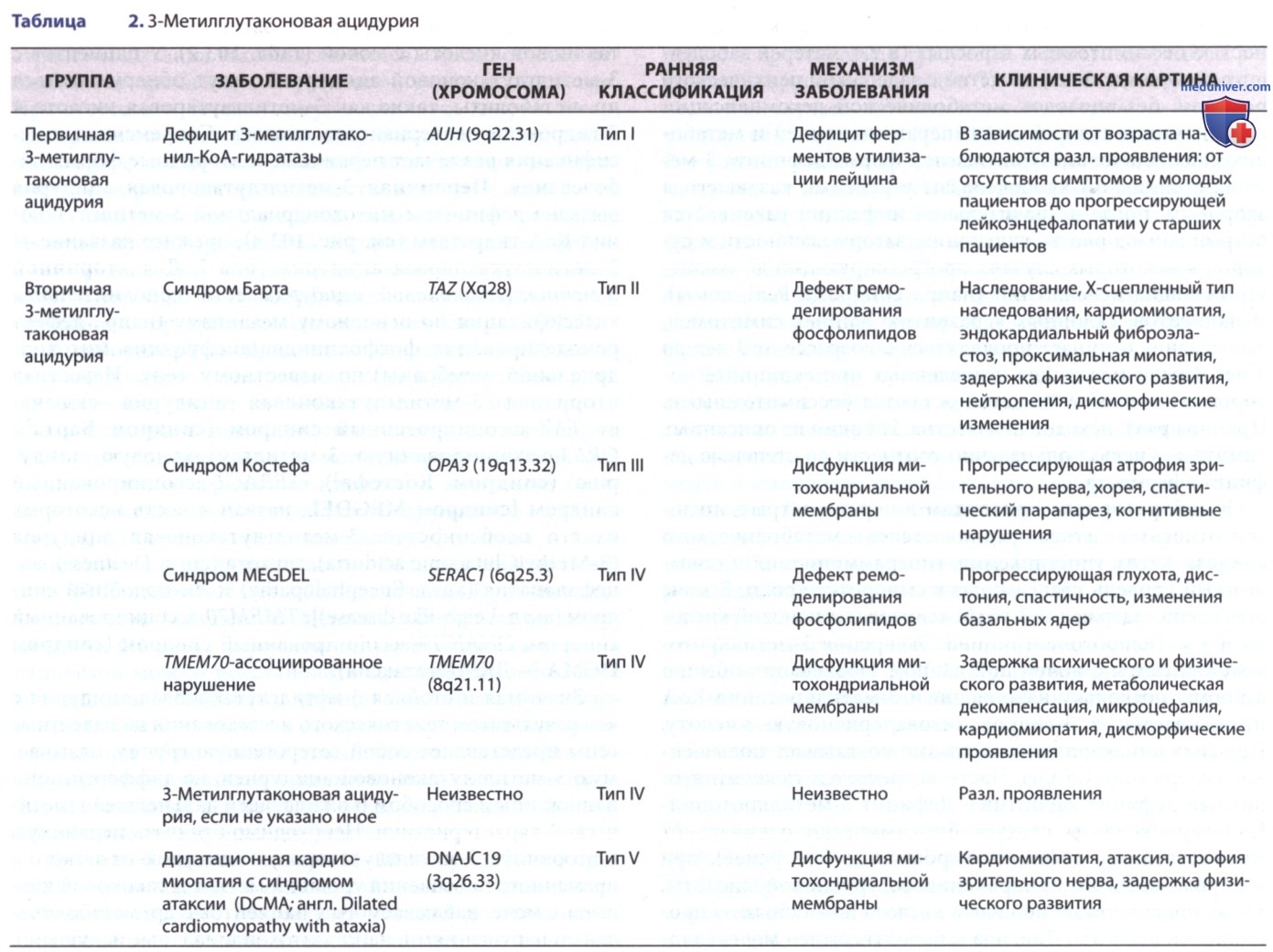
ж) 3-Метилглутаконовая ацидурия. 3-Метилглутаконовая ацидурия представляет собой гетерогенную группу метаболических нарушений, характеризующихся чрезмерной экскрецией 3-метилглутаконовой кислоты с мочой (табл. 2). У пациентов с 3-метилглутаконовой ацидурией могут обнаруживаться др. метаболиты, такие как 3-метилглутаровая кислота и 3-гидроксиизовалериановая кислота. Современная классификация различает первичные и вторичные формы заболевания. Первичная 3-метилглутаконовая ацидурия вызвана дефицитом митохондриальной 3-метилглутако-нил-КоА-гидратазы (см. рис. 4), прежнее название — 3-метилглутаконовая ацидурия типа I. Для вторичной 3-метилглутаконовой ацидурии есть дополнительная классификация по основному механизму (напр., дефект ремоделирования фосфолипидов/дисфункция митохондриальной мембраны)/по известному гену.
Известная вторичная 3-метилглутаконовая ацидурия включает TAZ-ассоциированный синдром (синдром Барта (Barth)); ОРАЗ-ассоциированную 3-метилглутаконовую ацидурию (синдром Костефа (Costeff)); SERAC1-ассоциированный синдром [синдром MEGDEL, назван в честь некоторых из его особенностей: 3-метилглутаконовая ацидурия (3-MEthylGlutaconic aciduria), глухота (англ. Deafness), энцефалопатия (англ. Encephalopathy) и Ли-подобный синдром (англ. Leigh-like disease)]; ТМЕМ70-ассоциированный синдром, DNAJC 19-ассоциированный синдром (синдром DCMA — ДКМП+атаксия).
Значимая и стойкая 3-метилглутаконовая ацидурия с «-» результатом генетического исследования на известные гены представляет собой гетерогенную группу, называемую 3-метилглутаконовой ацидурией, не дифференцированной иным способом и ожидающей дальнейшей генетической характеристики. Необходимо отличать первичную и вторичную 3-метилглутаконовую ацидурию от легкого и временного повышения уровня 3-метилглутаконовой кислоты в моче, наблюдаемого у пациентов с др. метаболическими нарушениями, напр. митохондриальные нарушения разл. этиологии.
1. Дефицит 3-метилглутаконил-КоА-гидратазы. Были описаны две основные клинические формы дефицита 3-метилглутаконил-КоА-гидратазы (см. рис. 4). При форме, присущей детскому возрасту, могут присутствовать неспецифические нарушения неврологического развития, такие как задержка/регресс речи, хореоатетоидные движения, атрофия зрительного нерва и задержка психомоторного развития средней степени тяжести. При повышенном катаболизме может возникнуть метаболический ацидоз. При взрослой форме пациенты могут не иметь клинических проявлений до 20-30 лет, но затем возникает клиническая картина медленно прогрессирующей лейкоэнцефалопатии с атрофией зрительного нерва, дизартрией, атаксией, спастичностью и деменцией. МРТ ГМ обычно выявляет аномалии белого в-ва, которые на годы могут предшествовать появлению клинических симптомов. Сообщалось также о бессимптомных детях и взрослых пациентах.
В моче пациентов наблюдается экскреция большого количества 3-метилглутаконовой кислоты и умеренного количества 3-гидроксиизовалериановой и 3-метилглутаровой кислот. Некоторым пациентам может помочь терапия L-карнитином. Эффективность диеты с низким содержанием лейцина не установлена. Заболевание наследуется по АуР-типу. Ген фермента гидратазы (AUH) расположен в хромосоме 9q22.31.
2. Синдром Барта (TAZ-ассоциированное расстройство). Это Х-сцепленное заболевание вызвано дефицитом тафаззина — митохондриального белка, кодируемого геном TAZ. Указанный фермент необходим для преобразования незрелого кардиолипина в его зрелую форму. Кардиолипин — митохондриальный фосфолипид, критически важный для обеспечения целостности внутренней митохондриальной мембраны. Клинические проявления синдрома Барта, которые обычно возникают на первом году жизни у мальчиков, включают кардиомиопатию, гипотонию, задержку роста, гипогликемию и нейтропению от легкой до тяжелой степени. Клинические симптомы могут проявляться во взрослом возрасте, но в большинстве случаев это происходит уже в подростковом возрасте. Если больные выживают в младенчестве, с возрастом может наступить относительное улучшение. Когнитивное развитие обычно в норме, хотя возможны задержка моторного развития и проблемы с обучаемостью.
К лабораторным признакам относится небольшое/умеренное увеличение экскреции с мочой 3-метилглутаконовой, 3-метилглутаровой и 2-этилгидракриловой кислот. В отличие от первичной 3-метилглутаконовой ацидурии (тип I), повышенной экскреции с мочой 3-гидроксиизова-лериановой кислоты не наблюдается. Активность фермента 3-метилглутаконил-КоА-гидратазы остается в норме. Часто наблюдается нейтропения. У некоторых пациентов наблюдались лактат-ацидоз, гипогликемия, низкая концентрация ХС в сыворотке, низкий уровень преальбумина и аномальная ультраструктура митохондрий. В культивированных фибробластах кожи данных пациентов очень низкое содержание общего кардиолипина и подклассов кардиолипина. В диагностике пациентов с «-»/неоднозначными результатами генетического анализа может помочь соотношение монолизокардиолипина/кардиолипина в культуре фибробластов. Из-за неспецифических проявлений заболевание, вероятно, не всегда диагностируется и не всегда регистрируется.
Заболевание наследуется как рецессивный признак, сцепленный с Х-хромосомой. Ген (TAZ) расположен в хромосоме Xq28. Умеренная 3-метилглутаконовая ацидурия, наблюдаемая при синдроме Барта, вероятно, вызвана дефектом митохондриальной мембраны, при котором происходит высвобождение данной органической кислоты. Специфического лечения не разработано. При кардиомиопатии пациентам с неудовлетворительным ответом на лечение может помочь трансплантация сердца. Для снижения риска инсульта предложен ежедневный прием ацетилсалициловой кислоты («Аспирина»).
3. ОРА-3-ассоциированная-3-метилглутаконовая ацидурия (синдром Костефа). У пациентов с синдромом Костефа наблюдаются такие клинические проявления, как ранняя атрофия зрительного нерва и более позднее развитие хореоатетоидных движений, спастичность, атаксия, дизартрия и когнитивные нарушения. В моче пациентов наблюдается экскреция умеренного количества 3-метилглутаконовой и 3-метилглутаровой кислот. Активность фермента 3-метилглутаконил-КоА-гидратазы остается в норме. Заболевание наследуется по АуР-типу. Ген данного заболевания (ОРА3) расположен в хромосоме 19q13.32. Предполагается, что мутации в ОРАЗ вызывают дисфункцию цепи транспорта электронов. Лечение симптоматическое.
4. Нарушения, прежде называемые 3-метилглутаконовой ацидурией типа IV. 3-Метилглутаконовая ацидурия типа IV представляет собой группу заболеваний с разнообразной генетической этиологией. Для двух заболеваний данной группы была обнаружена взаимосвязь с определенными генами, а молекулярные дефекты др. заболеваний еще не открыты.
Синдром MEGDEL (3-метилглутаконовая ацидурия с глухотой, энцефалопатией и Ли-подобным синдромом) — АуР-заболевание, вызванное патологическими мутациями в SERAC1 в хромосоме 6q25.3. У больных наблюдается прогрессирующая глухота, дистония, спастичность и повреждение базальных ядер, схожее с таковыми у пациентов с синдромом Ли. Лечение симптоматическое.
TMEM70-ассоциированное нарушение также наследуется по АуР-типу. Мутации в ТМЕМ70 вызывают дефицит митохондриального комплекса V, хотя точный механизм заболевания неизвестен. Клиническая симптоматика включает задержку и регресс психического развития, эпизоды, подобные синдрому Рея, умственную отсталость, задержку физического развития, микроцефалию, кардиомиопатию и дисморфические проявления. Пациенты склонны к метаболической декомпенсации, характеризующейся гипераммониемией (до 900 мкмоль/л) и лактатацидозом, которые чаще встречаются в 1-й год жизни. Острые эпизоды гипераммониемии лечат в/в введением декстрозы («Глюкозы»), липидной эмульсии, аммиак-захватывающими ЛП и в некоторых случаях гемодиализом. Описанная длительная терапия включает L-карнитин, кофермент Q10 и заместительную терапию бикарбонатами (напр., лимонной кислотой/натрия цитрат). Для ранней диагностики и лечения кардиомиопатии пациентам требуется периодическое проведение ЭхоКГ и ЭКГ.
5. DCMA-синдром (DNAJC19-ассоциированный синдром 3-метилглутаконовой ацидурии V типа). Синдром DCMA (ДКМП с атаксией) — это новое АуР-заболевание, выявленное у потомков канадских гуттеритов Дариуслейтов, живущих на плато Великие равнины Северной Америки. Как следует из сокращенного названия заболевания, у больных наблюдается ДКМП, удлиненный Q-Tc и поражение ЦНС. К неврологическим симптомам относятся умственная отсталость, поражение мозжечка и атрофия зрительного нерва. У всех пациентов нарушен рост. У 50% пациентов наблюдается ЗВУР. У заболевших мальчиков часто наблюдается крипторхизм и гипоспадия. Анализ мочи на органические кислоты показывает повышенное содержание 3-метилглутаконовой и 3-метилглутаровой кислот. Основной причиной синдрома DCMA являются мутации в DNAJC19 (3q26.33). Лечение симптоматическое. Периодические ЭхоКГ и ЭКГ позволяют проспективно выявить пациентов, нуждающихся в лечении ДКМП и удлиненного Q-Tc.
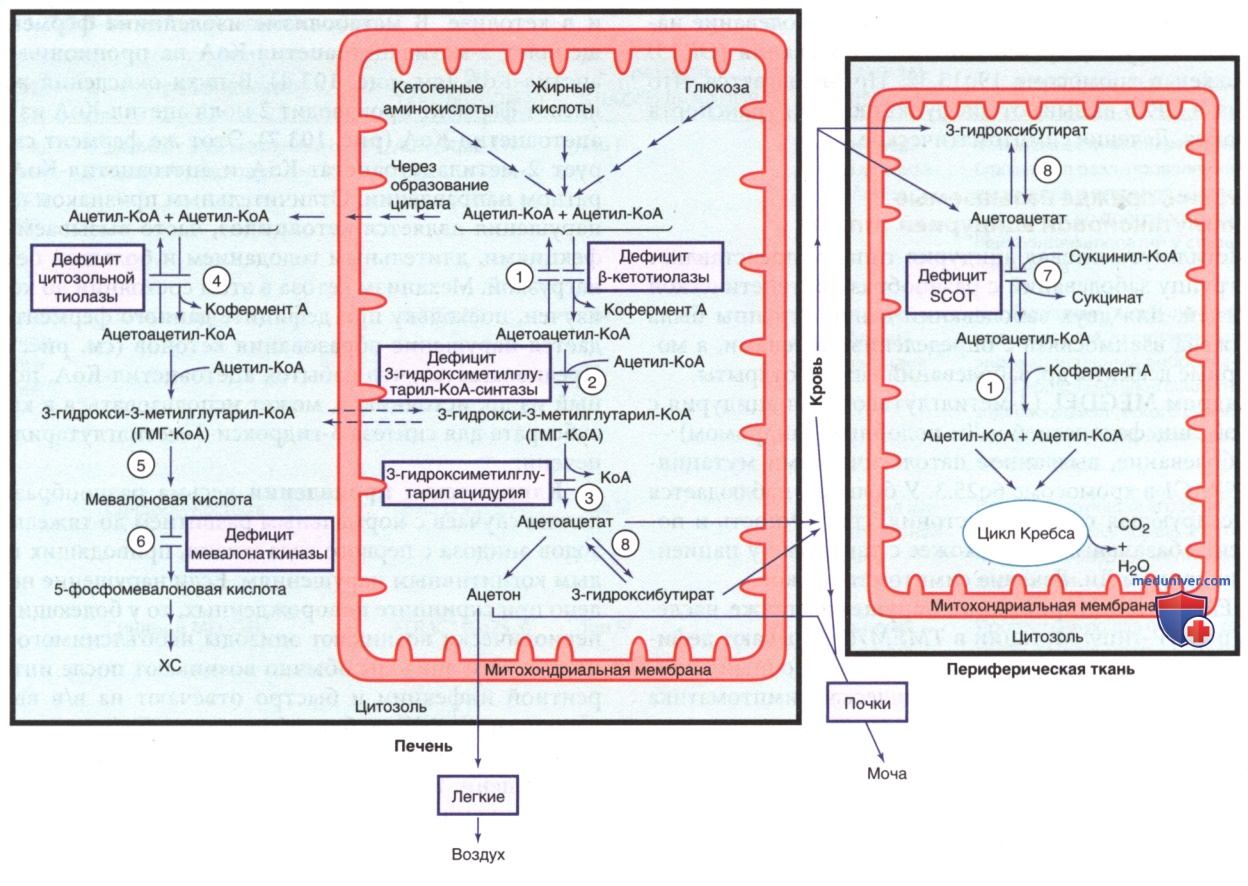
з) Дефицит β-кетотиолазы (3-оксотиолазы) [дефицит митохондриальной ацетоацетил-КоА-тиолазы (Т2)]. Указанный обратимый митохондриальный фермент участвует в последних стадиях катаболизма изолейцина и в кетолизе. В метаболизме изолейцина фермент расщепляет 2-метилацетоацетил-КоА на пропионил-КоА и ацетил-КоА (см. рис. 4). В пути окисления жирных кислот фермент производит 2 моля ацетил-КоА из 1 моля ацетоацетил-КоА (рис. 7). Этот же фермент синтезирует 2-метилацетоацетат-КоА и ацетоацетил-КоА в обратном направлении. Отличительным признаком данного нарушения является кетоацидоз, часто вызываемый инфекциями, длительным голоданием и большой белковой нагрузкой. Механизм кетоза в этом состоянии до конца не изучен, поскольку при дефиците данного фермента ожидается нарушение образования кетонов (см. рис. 7). Предполагается, что избыток ацетоацетил-КоА, полученный из др. источников, может использоваться в качестве субстрата для синтеза З-гидрокси-З-метилглутарил-КоА в печени.
Клинические проявления весьма разнообразны: от легких случаев с нормальным развитием до тяжелых эпизодов ацидоза с первого года жизни, приводящих к тяжелым когнитивным нарушениям. Если нарушение не выявлено при скрининге новорожденных, то у болеющих детей периодически возникают эпизоды необъяснимого кетоацидоза. Эти эпизоды обычно возникают после интеркуррентной инфекции и быстро отвечают на в/в введение жидкости и терапию бикарбонатами. Во время приступов может наблюдаться гипераммониемия легкой/умеренной степени. В единичных случаях сообщалось о гипо- и гипергликемии. В перерывах между данными эпизодами у ребенка симптомы могут полностью отсутствовать, и он может нормально переносить белковую диету. Когнитивные функции у большинства детей развиваются нормально. Приступы м.б. ошибочно верифицированы как отравление салицилатами, поскольку их клинические симптомы схожи, а повышенное содержание ацетоацетата в крови искажает результаты колориметрического анализа салицилатов.
К лабораторным признакам острого приступа относятся кетоацидоз и гипераммониемия. Наличие кетонов в моче и гипергликемия м.б. интерпретированы как ДКА; чтобы распознать данное метаболическое нарушение нужно обладать высокой степенью настороженности. Установить правильный диагноз позволяет анализ органических кислот в моче. В моче содержится большое количество 2-метилацетоацетата и его декарбоксилированных продуктов (бутанона, 2-метил-З-гидроксибутирата и тиглилглицина). Если пациенты находятся в стабильном состоянии, то в моче могут присутствовать более низкие концентрации метаболитов. Также вероятна легкая форма гиперглицинемии. Профиль ацилкарнитина в плазме показывает повышение содержания С5:1 и С5-ОН карнитинов, хотя концентрации данных метаболитов могут нормализоваться в перерывах между катаболическими эпизодами. При прохождении скрининга детьми с данной патологией без клинических проявлений на момент сбора анализа крови могут возникать л/о результаты из-за минимально повышенного уровня С5:1 и С5-ОН карнитинов.
Клинические и биохим. симптомы следует отличать от таковых при пропионовой и метилмалоновой ацидемии (см. ниже).
Лечение острых эпизодов направлено на восполнение потери жидкости. Резистентный к лечению метаболический ацидоз м.б. достаточно тяжелым, в этом случае требуется инфузионное введение бикарбоната. Для подавления кетоза, липолиза и кетогенеза используется 10% р-р декстрозы («Глюкозы») с соответствующими электролитами. При длительной терапии рекомендуется ограничить потребление белка в соответствии с возрастными физиол. нормами. Также рекомендуется прием L-карнитина внутрь (50-100 мг/кг в сутки) для профилактики возможного вторичного дефицита карнитина. Долгосрочный прогноз возможного нормального качества жизни, по всей видимости, очень благоприятный. Зарегистрирован случай успешной беременности с нормальным исходом.
Дефицит β-кетотиолазы наследуется по АуР-типу и, возможно, встречается чаще, чем это предполагалось ранее. Ген (АСАТ1) данного фермента расположен в хромосоме 11q22.3. Диагноз м.б. подтвержден молекулярным анализом гена ACAT1/исследованием активности фермента в лейкоцитах/культуре фибробластов.
и) Дефицит цитозольной ацетоацетил-КоА-тиолазы. Указанный фермент катализирует синтез ацетоацетил-КоА из 2 моля ацетил-КоА в цитозоле (см. рис. 7). Ацетоацетил-КоА в цитозоле является предшественником синтеза ХС в печени. Цитозольную ацетоацетил-КоА-тиолазу следует отличать от митохондриальной тиолазы (см. ранее и рис. 4). Клинические проявления у пациентов с этим очень редким дефицитом фермента описаны не полностью. У пациентов в первые несколько месяцев жизни могут наблюдаться прогрессирующая задержка психического развития в тяжелой форме, гипотония и хореоатетоидные движения. Лабораторные признаки неспецифичны: в крови и моче м.б. повышены уровни лактата, пирувата, ацетоацетата и 3-гидроксибутирата. Описан случай, когда у пациента уровни ацетоацетата и 3-гидроксибутирата были нормальными. Диагностика м.б. основана на выявлении дефицита активности цитозольной тиолазы при биопсии печени/в культуре фибробластов и методом ДНК-анализа. Эффективное лечение неизвестно, хотя диета с низким содержанием жиров помогла уменьшить кетоз у одного пациента.
к) Дефицит митохондриальной 3-гидрокси-3-метилглутарил-КоА-синтазы. Указанный фермент катализирует синтез 3-гидрокси-3-метилглутарил-КоА из ацетоацетил-КоА и ацетил-КоА в митохондриях. Это ключевой этап в синтезе кетоновых тел в печени (см. рис. 7). Зарегистрировано всего несколько случаев дефицита данного фермента у пациентов. Основным клиническим синдромом является гипокетотическая гипогликемия, вызванная физиол. стрессом, напр. инфекциями/голоданием. Возраст на момент обращения варьировал от младенчества до 6 лет. Как правило, до этих эпизодов у детей не проявляется никаких симптомов и при соответствующем лечении после выздоровления их состояние может оставаться стабильным (за исключением легкой гепатомегалии с липодистрофией). Последующие эпизоды можно предотвратить, избегая длительного голодания при последующих интеркуррентных заболеваниях.
Гепатомегалия является постоянным признаком у данных пациентов. К лабораторным признакам относятся гипогликемия, ацидоз с/без легкого кетоза, повышенные уровни печеночных проб и массивная дикарбоновая ацидурия. Клинические и лабораторные признаки можно ошибочно принять за нарушения метаболизма жирных кислот. У пациентов с дефицитом 3-гидрокси3-метилглутарил-КоА-синтазы, в отличие от пациентов с нарушением окисления жирных кислот, конъюгаты ацилкарнитина в крови отсутствуют. Лечение вторичного дефицита карнитина приемом БАД L-карнитина может привести к повышению ацетилкарнитина (С2-карнитина) в плазме, что, вероятно, обусловлено в/клеточным накоплением ацетил-КоА. Контрольное исследование натощак позволяет выявить клинические и биохим. признаки.
Лечение заключается в обеспечении достаточным количеством калорий и предотвращении продолжительных периодов голодания. Соблюдение диеты с ограничением белка не требовалось.
Заболевание наследуется по АуР-типу. Ген (HMGCS2) данного фермента расположен в хромосоме 1р12. Любого ребенка с гипокетотической гипогликемией натощак следует проверять на это заболевание, оно может встречаться чаще, чем предполагается.
л) Дефицит 3-гидрокси-3-метилглутарил-КоА-лиазы (3-гидрокси-3-метилглутаровая ацидурия). 3-Гидрокси-3-метилглутарил-КоА-лиаза (см. рис. 4) катализирует превращение 3-гидрокси-3-метилглутарил-КоА в ацетоацетат и является ферментом, ограничивающим скорость кетогенеза (см. рис. 7). Дефицит данного фермента представляет собой редкое нарушение, встречающееся с повышенной частотой в Саудовской Аравии, на Пиренейском полуострове и в Бразилии у пациентов португальского происхождения. Клинически у 30% пациентов симптомы появляются в первые несколько дней жизни, а у >60% — в 3-11 мес. В редких случаях пациенты остаются бессимптомными до подросткового возраста. Если определение дефицита 3-гидрокси3-метилглутарил-КоА-лиазы с использованием С5-ОН-карнитина включено в скрининг новорожденных, многие младенцы выявляются в период новорожденности до появления симптомов. Подобно дефициту 3-гидрокси-3-метилглутарил-КоА-синтазы, пациенты с дефицитом 3-гидрокси-3-метилглутарил-КоА-лиазы могут страдать от острой гипокетотической гипогликемии.
Эпизоды рвоты, тяжелой гипогликемии, гипотонии, ацидоза с кетозом или без него и обезвоживание могут быстро привести к заторможенности, атаксии и коме. Такие эпизоды часто возникают при повышенном катаболизме, напр. длительном голодании/интеркуррентной инфекции. Часто наблюдается гепатомегалия. Эти проявления м.б. ошибочно приняты за синдром Рея/нарушения окисления жирных кислот, напр. за дефицит среднецепочечной ацил-КоА-дегидрогеназы. Отдаленные последствия могут включать ДКМП, стеатоз печени и панкреатит. Уровень развития м.б. в пределах нормы, но после длительных эпизодов гипогликемии у пациентов наблюдались умственная отсталость и судороги с аномалиями белого в-ва на МРТ.
К лабораторным признакам относятся гипогликемия, умеренная и тяжелая гипераммониемия и ацидоз. Кетоз умеренный/отсутствует (см. рис. 7). Экскреция с мочой 3-гидрокси-3-метилглутаровой кислоты и др. близких промежуточных метаболитов катаболизма лейцина (3-метилглутаровой кислоты, 3-метилглутаконовой кислоты и 3-гидроксиизовалериановой кислоты) значительно увеличивается, из-за чего та приобретает запах кошачьей мочи. В моче в период острых приступов также м.б. повышено содержание глутаровой и дикарбоновой кислот. Часто обнаруживают вторичный дефицит карнитина. Заболевание наследуется как АуР-признак. 3-Гидрокси-3-метилглутарил-КоА-лиаза кодируется геном HMGCL. Диагноз м.б. подтвержден молекулярным исследованием гена HMGCL/исследованием активности фермента в культуре фибробластов, лейкоцитах и биоптатах печени. Пренатальную диагностику можно выполнить методом молекулярного ДНК-анализа при известных семейных мутациях/исследованием активности фермента в культуре амниоцитов/биопсии ворсинок хориона.
Лечение острых эпизодов включает восполнение жидкости организма, инфузию декстрозы («Глюкозы») для контроля гипогликемии, обеспечение адекватным калоражем при кормлении и введение бикарбонатов для коррекции ацидоза. Лечение гипераммониемии следует начинать незамедлительно. Пациентам с тяжелой гипераммониемией, трудно поддающейся лечению, может потребоваться заместительная почечная терапия. В качестве долгосрочного лечения рекомендуется диета с ограниченным потреблением белков и жиров. Прием L-карнитина внутрь (50-100 мг/кг в сутки) предотвращает вторичный дефицит карнитина. Следует избегать длительного голодания.
м) Дефицит сукцинил-КоА:3-оксоацид-КоА-трансферазы. Дефицит сукцинил-КоА:3-оксоацид-КоА-трансферазы и дефицит β-кетотиолазы вместе определяются как нарушения утилизации кетонов. Сукцинил-КоА:3-оксоацид-КоА-трансфераза участвует в превращении кетоновых тел (ацетоацетат и 3-гидроксибутират), образующихся в митохондриях печени, в ацетоацетил-КоА в непеченочных тканях (см. рис. 7). Дефицит данного фермента приводит к накоплению кетоновых тел, кетоацидозу, повышенному расходу глюкозы и гипогликемии. При голодании у пациентов обычно пропорционально увеличивается количество свободных жирных кислот в плазме. В настоящее время зарегистрировано >30 пациентов с дефицитом данного фермента. Вероятно, заболевание не редкое, поскольку многие случаи могут проходить в легкой форме и оставаться недиагностированными. При дефиците сукцинил-КоА:3-оксоацид-КоА-трансферазы отсутствуют 2-метилацетоацетат, 2-метил-З-гидроксибутират и тиглилглицин, характерные для дефицита β-кетотиолазы, что позволяет различить эти два заболевания.
Профиль ацилкарнитина в плазме обычно не содержит каких-либо специфических изменений.
Обычно острый эпизод тяжелого кетоацидоза возникает у ребенка, который рос и развивался нормально. Симптомы проявляются в первую неделю жизни у ~ 1/2 пациентов и практически у всех <2 лет. Острый эпизод часто провоцируется состоянием, обуславливающим повышенный катаболизм: инфекция/длительное голодание. При отсутствии лечения эпизод кетоацидоза может привести к смерти. В межприступный период может сохраняться хронический субклинический кетоз. Обычно ребенок развивается нормально, хотя тяжелые и повторяющиеся эпизоды кетоацидоза и гипогликемии могут предрасполагать к нейрокогнитивным нарушениям.
Лабораторные признаки во время острого эпизода неспецифичны и включают метаболический ацидоз и кетонурию с повышенным содержанием ацетоацетата и 3-гидроксибутирата в крови и моче. Никаких др. органических кислот в крови/моче не обнаруживается. Содержание глюкозы в крови обычно в норме, однако были зарегистрированы случаи гипогликемии у некоторых новорожденных с тяжелым кетоацидозом. Аминокислоты и ацилкарнитиновый профиль плазмы обычно в норме. Тяжелая форма дефицита сукцинил-КоА:3-оксоацид-КоА-трансферазы может сопровождаться кетозом, даже если пациенты клинически стабильны. Любого ребенка с необъяснимыми приступами кетоацидоза следует проверить на это заболевание. Диагноз м.б. установлен при генетическом исследовании ОХСТ1/определении дефицита активности фермента в культуре фибробластов. Заболевание наследуется по АуР-типу.
Лечение острых эпизодов включает восстановление водного баланса р-рами декстрозы («Глюкозы»), коррекцию ацидоза и соблюдение диеты с необходимым содержанием калорий. Долгосрочное лечение должно включать соблюдение высокоуглеводной диеты, предотвращение длительного голодания и введение декстрозы («Глюкозы») до ожидаемого или во время установившегося катаболического состояния.
н) Дефицит мевалонаткиназы. Мевалоновая кислота, промежуточный метаболит синтеза ХС, превращается в 5-фосфомевалоновую кислоту под действием фермента мевалонаткиназы (см. рис. 7). По клиническим проявлениям и степени дефицита фермента различают два заболевания: мевалоновую ацидурию и синдром гипериммуноглобулинемии D. Оба заболевания сопровождаются рекуррентной лихорадкой, желудочно-кишечными симптомами, изменениями кожи и слизистых оболочек и лимфаденопатией. У пациентов с мевалоновой ацидурией наблюдаются замедление темпов роста и поражение НС. На практике эти два заболевания располагаются на двух противоположных полюсах спектра.
1. Мевалоновая ацидурия. К клиническим проявлениям относятся задержка физического развития, замедление темпов роста, умственная отсталость, гипотония, атаксия, миопатия, гепатоспленомегалия, катаракта и дисморфизм лица (долихоцефалия, выступающие лобные бугры, низко расположенные ушные раковины, направленный вниз разрез глаз, длинные ресницы). У большинства пациентов возникают периодические кризисы, характеризующиеся лихорадкой, рвотой, диареей, гепатоспленомегалией, артралгией, лимфаденопатией, отеками и кореподобной сыпью. Эти эпизоды обычно длятся 2-7 сут и повторяются до 25 р/год. Кризисы могут привести к летальному исходу.
Лабораторным признаком является значительное повышение содержания мевалоновой кислоты в моче. Концентрация мевалоновой кислоты в моче 500-56 ООО ммоль/моль креатинина (в норме <0,3 ммоль/моль креатинина). Также значительно повышается содержание мевалоновой кислоты в плазме (до 540 мкмоль/л; в норме <0,04 мкмоль/л). Содержание мевалоновой кислоты, как правило, коррелирует с тяжестью состояния и повышается в период кризисов. Концентрация ХС в сыворотке остается в норме/незначительно снижается. Концентрация КФК в сыворотке может значительно увеличиться. В период кризисов повышается уровень маркеров воспаления. При проведении МРТ ГМ м.б. выявлена прогрессирующая атрофия мозжечка.
Диагноз подтверждается анализом ДНК и анализом активности мевалонаткиназы в лимфоцитах/культуре фибробластов. Активность ферментов при данной форме заболевания ниже порога обнаружения. В период острых кризисов лечение преднизоном в высоких дозах помогает, но в течение длительного времени его обычно не применяют из-за побочных эффектов. Этанерцепт (ингибитор ФНО) и анакинра (антагонист рецептора IL-1) оказались эффективными и позволяют достигать значительных клинических улучшений. Заболевание наследуется по АуР-типу. Возможно проведение пренатальной диагностики путем выявления установленных родственных мутаций MVK, измерения мевалоновой кислоты в амниотической жидкости и анализа активности фермента в культуре ам-ниоцитов/образцах ворсинок хориона. Ген (MVK) данного фермента расположен в хромосоме 12q24.11.
2. Синдром гипериммуноглобулинемии D (гипериммуноглобулинемия D и синдром рекуррентной лихорадки). Некоторые мутации гена мевалоновой киназы (MVK) вызывают дефицит ферментов более легкой степени, что клинически проявляется рекуррентной лихорадкой с гипериммуноглобулинемией D. У таких пациентов наблюдаются периодические приступы лихорадки, сопровождаемые болями в животе, рвотой, диареей, артралгией, артритом, гепатоспленомегалией, лимфаденопатией и кореподобной сыпью (даже петехии и геморрагическая сыпь), которые обычно дебютируют в 1-й год жизни. Приступы м.б. спровоцированы вакцинацией, незначительной травмой и стрессом и могут рецидивировать каждые 1-2 мес и длиться по 2-7 дней. В перерывах между острыми приступами симптомы отсутствуют. Диагностическим лабораторным признаком является повышение уровня сывороточного IgD. У 80% пациентов также повышен IgA. Во время острых приступов возможны лейкоцитоз, повышенный уровень СРВ и легкая форма мевалоновой ацидурии. Высокие концентрации сывороточного IgD позволяют провести ДД этого заболевания с семейной средиземноморской лихорадкой.
Рекомендации по лечению приведены в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.
о) Пропионовая ацидемия (дефицит пропионил-КоА-карбоксилазы). Пропионовая кислота представляет собой промежуточный метаболит изолейцина, валина, треонина, метионина, жирных кислот с нечетным числом атомов углерода в молекуле и боковых цепей ХС. Обычно пропионовая кислота в форме пропионил-КоА подвергается карбоксилированию до D-метилмалонил-КоА. Процесс катализируется митохондриальным ферментом пропионил-КоА-карбоксилазой. Этому ферменту биотин необходим в качестве кофактора; т.о., нарушения метаболизма биотина, среди прочего, могут также приводить к повышению содержания метаболитов пропионовой кислоты (см. рис. 4). Пропионил-КоА-карбоксилаза представляет собой мультимерный фермент, состоящий из двух неидентичных субъединиц, α и β, кодируемых двумя генами, РССА и РССВ соответственно. Мутации пропионил-КоА-карбоксилазы вызывают нарушение, называемое пропионовой ацидемией.
Клинические признаки пропионовой ацидемии неспецифичны. Симптомы появляются в тяжелой форме в первые дни жизни пациентов. Затруднения вскармливания, рвота, гипотония, заторможенность, обезвоживание, сепсисоподобное состояние и клинические признаки тяжелого кетоацидоза быстро прогрессируют до комы и смерти. У 30% младенцев с данной патологией случаются кризы. Если у ребенка развился первый приступ, похожие эпизоды метаболической декомпенсации могут проявляться в период интеркуррентной инфекции, травмы, операции, длительного голодания, длительного запора, после приема пищи с высоким содержанием белка. У переживших это состояние часто наблюдаются умственная отсталость (средней/тяжелой степени) и неврологические признаки, соответствующие экстрапирамидной (дистония, хореоатетоз, тремор) и пирамидной (параплегия) дисфункциям. При нейровизуализации очевидно, что данные аномалии, часто возникающие после метаболического криза, являются результатом повреждения базальных ядер, особенно бледного шара. Данное явление получило название метаболического инсульта. Это основная причина неврологических осложнений у детей, переживших приступ пропионовой ацидемии.
К дополнительным долгосрочным осложнениям относятся задержка физического развития, атрофия зрительного нерва, панкреатит, кардиомиопатия и остеопения.
В более легкой форме эпизоды метаболического криза встречаются реже, но эти дети все еще подвержены риску развития умственной отсталости, судорог, удлиненного интервала Q-Tc и тяжелой кардиомиопатии. Универсальный скрининг новорожденных позволяет выявить пропионовую ацидемию путем определения повышенного содержания пропионилкарнитина (С3) в сухих пятнах крови. Однако у пациентов с легкой формой пропионовой ацидемии пропионилкарнитин может оставаться ниже порогового значения скрининга, установленного лабораторией, что приводит к л/о результату. Т.о., врачам необходимо сохранять высокий уровень настороженности в отношении данного заболевания и в дальнейшем проводить биохим. обследование младенцев и детей с необъяснимым кетозом/метаболическим ацидозом.
К лабораторным признакам в период острого приступа относят метаболический ацидоз разл. степени тяжести, часто с большой анионной разницей, кетоз, кетонурию, гипогликемию, анемию, нейтропению и тромбоцитопению. Тяжесть гипераммониемии обычно варьирует от умеренной до тяжелой. Концентрация аммиака в плазме обычно коррелирует с тяжестью заболевания. В отличие от др. причин гипераммониемии, концентрация глутамина в плазме обычно находится в пределах нормы/понижена. Наличие тяжелого метаболического ацидоза и нормального/пониженного содержания глутамина в плазме позволяет отличить пропионовую ацидемию от гипераммониемии, вызванной нарушениями цикла мочевины. Измерение аммиака в плазме особенно полезно при планировании терапевтической стратегии в период обострения у пациентов с установленным диагнозом.
Механизмы гипераммониемии при пропионовой ацидемии недостаточно изучены, но, вероятно, обусловлены нарушением биохим. и pH сред митохондриального матрикса, где находится проксимальная часть цикла мочевины. Концентрация глицина м.б. повышена во всех физиол. жидкостях (кровь, моча, СМЖ), что, возможно, является результатом ингибирования системы расщепления глицина в митохондриях печени (рис. 8). У пациентов с метилмалоновой ацидемией наблюдалось повышение уровня глицина. Эти нарушения раньше были объединены под общим названием кетотическая гиперглицинемия, но позже были выявлены дефициты конкретных ферментов. У таких пациентов может наблюдаться легкое/средней степени выраженности повышение уровня лактата и лизина в крови. В плазме и моче младенцев с пропионовой ацидемией значительно повышены концентрации пропионилкарнитина, 3-гидроксипропионовой и метилцитриновой кислот (предположительно образующейся в результате конденсации пропионил-КоА с щавелевоуксусной кислотой).
В моче могут присутствовать пропионилглицин и др. промежуточные метаболиты катаболизма аминокислот с разветвленной цепью (напр., тиглилглицин). В межприступный период в крови может сохраняться умеренное повышение уровня глицина и вышеуказанных органических кислот. Визуализация ГМ может выявить церебральную атрофию, миелинизацию с поздним началом и аномалии бледного шара (и др. частей базальных ядер).
Диагноз пропионовой ацидемии следует отличать от множественного дефицита карбоксилаз (см. выше и рис. 6). Кроме метаболитов пропионовой кислоты, у младенцев с множественным дефицитом карбоксилаз наблюдается экскреция молочной кислоты, 3-метилкро-тонилглицина и 3-гидроксиизовалериановой кислоты в большом количестве. Наличие гипераммониемии может указывать на генетическое нарушение ферментов, участвующих в цикле мочевины. У младенцев с нарушениями цикла мочевины обычно не наблюдается ацидоз (см. рис. 1) и обнаруживается повышенный уровень глутамина в плазме. Окончательный диагноз пропионовой ацидемии м.б. установлен на основании молекулярного анализа генов РССА и РССВ/измерением активности фермента в лейкоцитах и культуре фибробластов.
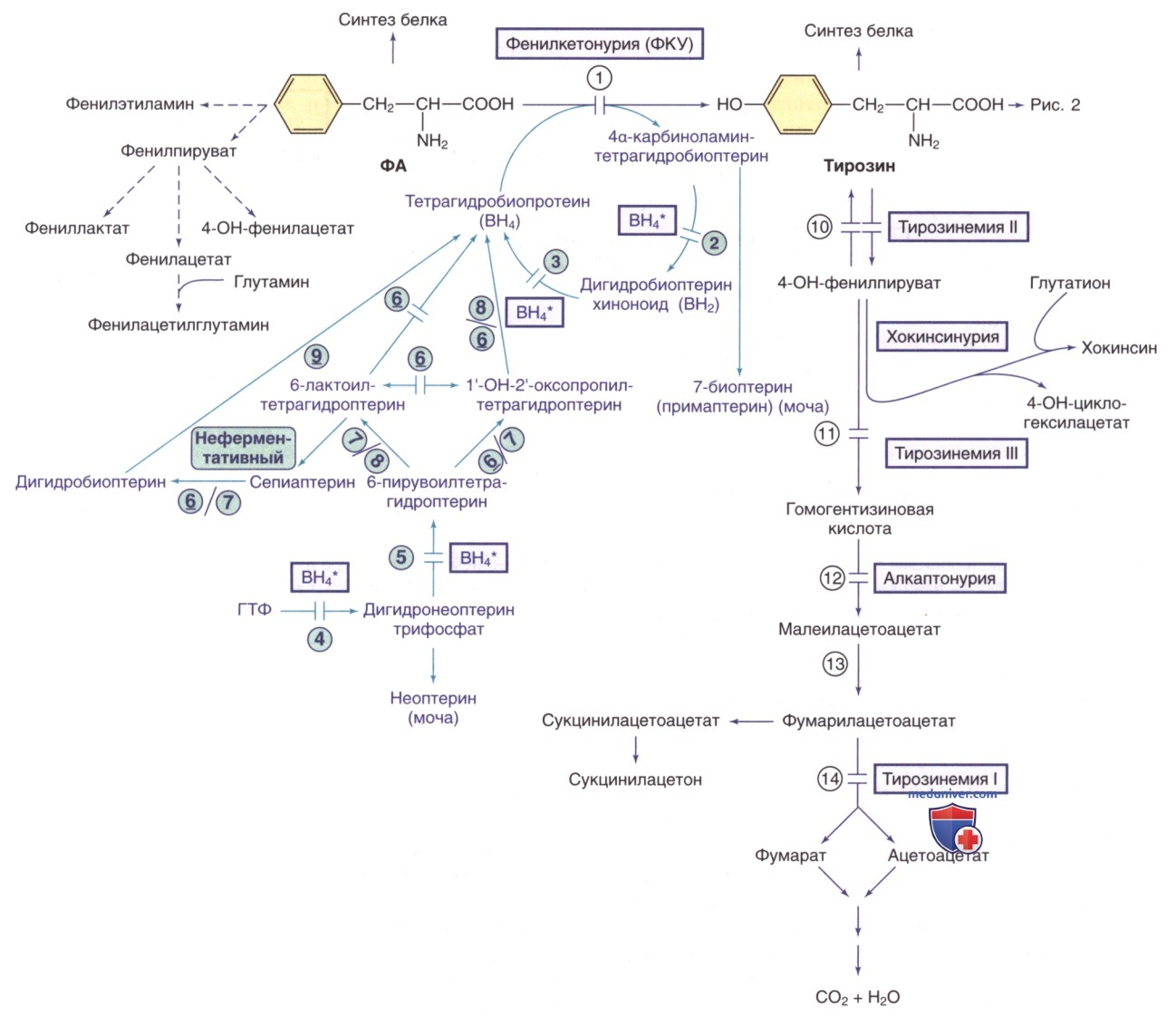
Рисунок 1. Пути метаболизма фенилаланина (ФА) и тирозина. Генетически детерминированные ферментативные нарушения показаны горизонтальными линиями, пересекающими стрелку(-и) реакции. Пути синтеза кофактора тетрагидробиоптерина (ВН4) показаны фиолетовым цветом. ВН4* относится к дефектам метаболизма ВН4, влияющим на гидроксилазы ФА, тирозина и триптофана (рис. ниже). Ферменты: 1) фенилаланингидроксилаза; 2) птеринкарбиноламиндегидратаза; 3) дигидробиоптеринредуктаза; 4) гуанозинтрифосфат (ГТФ)-циклогидролаза; 5) 6-пирувоилтетрагидроптеринсинтаза; 6) сепиаптеринредуктаза; 7) карбонилредуктаза; 8) альдолазоредуктаза; 9) дигидрофолатредуктаза; 10) тирозинаминотрансфераза; 11) 4-гидроксифенилпируватдиоксигеназа; 12) диоксигеназа гомогентизиновой кислоты; 13) малеилацетоацетат изомераза; 14) фумарилацетоацетат гидролаза
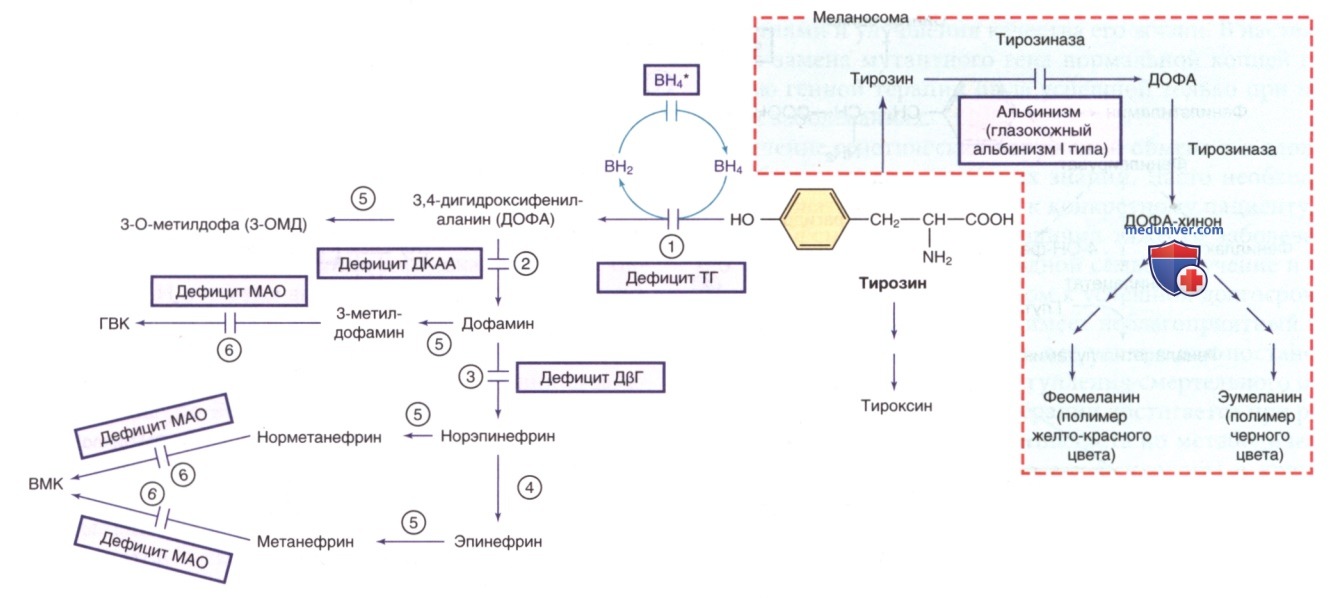
Другие пути метаболизма тирозина. ВН4* указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). ГВК — гомованиловая кислота; ВМК — ванилилминдальная кислота; ВН4 — тетрагидробиоптерин. Ферменты: 1) тирозингидроксилаза (ТГ); 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) дофамин-β-гидроксилаза (ДβГ); 4) фенилэтаноламин-N-метилтрансфераза (ФНМТ); 5) катехол-О-метилтрансфераза (КОМТ); 6) МАО
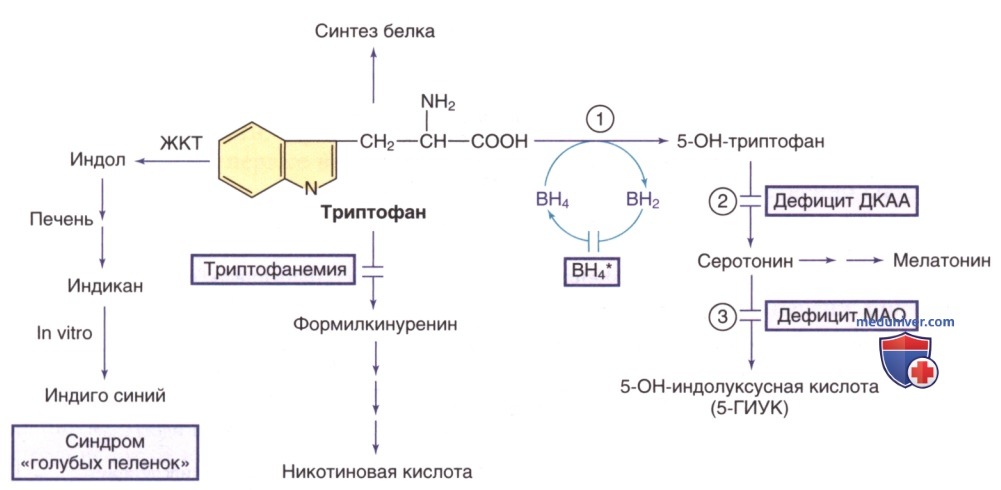
Пути метаболизма триптофана. Тетрагидробиоптерин (ВН4*) указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. ниже). Ферменты: 1) триптофангидроксилаза; 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) МАО
Лечение острых метаболических кризов включает восстановление водного баланса р-рами декстрозы («Глюкозы»), коррекцию ацидоза и купирование эпизодов повышенного катаболизма путем обеспечения адекватной по калоражу диеты, за счет усиленного энтерального/ парентерального питания. Часто необходимо ограничить потребление белка на короткий период (не >24 ч). В зависимости от клинического состояния рекомендуется постепенное повторное введение белка. Если пациент не переносит энтеральное кормление >48 ч ограничения белка, необходимо назначить парентеральное питание с учетом возраста для достижения рекомендованного потребления белка с пищей. Пациентам с непереносимостью рекомендуемой нормы потребления белка с пищей могут потребоваться специальные продукты лечебного питания, не содержащие изолейцин, валин, треонин и метионин. Состав и количество белка в диете у пациентов отличаются. Состав метаболической диеты можно регулировать, отслеживая уровень и количество аминокислот в плазме, взятых через 3-4 ч после обычного кормления. Некоторым пациентам может помочь подавление пропионогенной микрофлоры кишечника.
Этого можно добиться приемом антибактериальных ЛП внутрь, напр., неомицина/метронидазола.
Следует избегать длительного использования метронидазола, поскольку он вызывает обратимую периферическую невропатию и увеличение интервала Q-Tc. Риск удлинения Q-Tc м.б. проблемой у пациентов с пропионовой ацидемией, подверженных риску развития кардиомиопатии и удлинения интервала Q-T. До и после терапии метронидазолом рекомендуется выполнить стандартную ЭКГ и суточный монитор ЭКГ (холтер-мониторирование). Эффективным м.б. купирование запора.
У пациентов с пропионовой ацидемией часто развивается вторичный дефицит карнитина, предположительно в результате потери пропионилкарнитина с мочой. Введение L-карнитина (внутрь/в/в 50-100 мг/кг в сутки) способствует восстановлению свободного карнитина в крови. При лечении пациентов с сопутствующей гипераммониемией следует принимать меры по снижению уровня аммиака в крови. Пациентам в тяжелом состоянии, с ацидозом и гипераммониемией требуется гемодиализ для быстрого и эффективного удаления аммиака и др. токсичных соединений. В лечении острой гипераммониемии могут помочь карглумовая кислота и акцепторы азота (натрия бензоат, фенилацетат натрия, натрия фенилбутират). Хотя ни у одного младенца с пропионовой ацидемией не зафиксирован ответ на биотин, его необходимо применять (внутрь 10 мг/сут) у всех младенцев во время первого приступа до постановки диагноза и исключения множественного дефицита карбоксилаз.
Длительное лечение заключается в низкобелковой диете, соответствующей возрасту, и приеме L-карнитина (внутрь 50-100 мг/кг в сутки). В некоторых центрах лечение легких случаев пропионовой ацидемии осуществляют без продуктов лечебного питания, просто ограничивая потребление белка рекомендуемой дозой. Пациентам с непереносимостью рекомендуемой нормы потребления белка с пищей могут потребоваться специальные продукты лечебного питания, не содержащие предшественников пропионата (изолейцин, валин, метионин и треонин). Чрезмерное употребление продуктов лечебного питания при ограничении белка из натуральных источников может вызвать дефицит незаменимых аминокислот, особенно изолейцина и валина. В связи с этим может развиться состояние, напоминающее энтеропатический акродерматит. Чрезмерное ограничение метионина, особенно в первые годы жизни, может способствовать замедлению роста ГМ и развитию микроцефалии. Для предотвращения данной проблемы основным пищевым источником белка должны быть натуральные продукты питания.
Некоторым пациентам может потребоваться замещение бикарбонатов (напр., лимонной кислотой/цитратом натрия) для коррекции хронического ацидоза. В период между приступами концентрация аммиака в плазме обычно нормализуется, хотя у некоторых пациентов может наблюдаться легкая хроническая гипераммониемия. Острые приступы, вызванные инфекциями, голоданием, травмами, стрессом, запорами и неправильным питанием, следует лечить быстро и интенсивно. При соблюдении диеты требуется тщательный мониторинг аммиака и аминокислот (особенно изолейцина, лейцина, валина, треонина и метионина) в плазме крови, взятой через 3-4 ч после последнего приема пищи, а также контроль параметров роста. Клинически нестабильным пациентам с рецидивирующей гипераммониемией, частыми метаболическими кризами и замедленным ростом назначают ортотопическую трансплантацию печени. Однако она не излечивает пропионовую ацидемию, поэтому рекомендуется пожизненное соблюдение диеты и профилактическое лечение в периоды значительного метаболического стресса.
Долгосрочный прогноз делают с осторожностью. Острый приступ может привести к летальному исходу. При легкой форме заболевания, выявленного при скрининге новорожденных, возможно нормальное психомоторное развитие. У детей с диагнозом, установленным при клинической манифестации, в некоторой степени могут наблюдаться постоянные неврологические признаки (напр., тремор, дистония, хорея и спастичность), несмотря на лечение. Эти неврологические симптомы м.б. последствиями метаболического инсульта, возникшего во время острого метаболического приступа. Несмотря на достаточный метаболический контроль, у детей старшего возраста могут возникнуть удлиненный интервал Q-Tc, кардиомиопатия с возможным прогрессированием до СН, несовместимых с жизнью аритмий и летального исхода. Частым и тяжелым осложнением пропионовой ацидемии является острый панкреатит.
Остеопороз предрасполагает к переломам, которые могут возникнуть даже после незначительной механической нагрузки.
Возможно проведение пренатальной диагностики на основании выявления известных семейных мутаций в РССА/РССВ и методом измерения активности фермента в культивируемых амниотических клетках/образцах некультивируемых ворсинок хориона.
Пропионовая ацидемия наследуется по АуР-типу, частота встречаемости во всем мире составляет 1:105 000-250 000 живорожденных. Патология наиболее часто встречается у гренландских инуитов (1:1000) и у некоторых саудовских племен (1:2000-5000 живорождений). Ген α-субъединицы (РССА) расположен в хромосоме 13q32.3, ген β-субъединицы (РССВ) — в хромосоме 3q22.3. Мутации любого гена приводят к сходным клиническим и биохим. проявлениям. Несмотря на известные случаи беременностей с нормальным исходом, перинатальный период представляет особый риск для женщин с пропионовой ацидемией вследствие развития чрезмерной рвоты беременных (лат. hyperemesis gravidarum), декомпенсации кардиомиопатии, изменения потребности в белке и риска метаболической декомпенсации.
п) Изолированные метилмалоновые ацидемии. Метилмалоновые ацидемии представляют собой группу метаболических заболеваний разл. этиологии, характеризующихся нарушением превращения метилмалонил-КоА в сукцинил-КоА. Пропионил-КоА, полученный в результате катаболизма изолейцина, валина, треонина, метионина, боковой цепи ХС и жирных кислот с нечетным числом атомов углерода в молекуле, катализируется пропионил-КоА-карбоксилазой с образованием D-метилмалонил-КоА. Затем метилмалонил-КоА-эпимераза превращает D-метилмалонил-КоА в энантиомер L-метилмалонил-КоА. Дефицит метилмалонил-КоА-эпимеразы представляет собой редкое заболевание, характеризующееся стойким повышением уровня метаболитов пропионата и метилмалоновой кислоты. Данная патология может проявляться в виде метаболического ацидоза, кетоза, однако пациенты с дефицитом метилмалонилКоА-эпимеразы кажутся клинически стабильнее, нежели пациенты с тяжелыми формами метилмалоновой ацидемии.
На следующем биохим. этапе метилмалонил-КоА-мутаза способствует превращению L-метилмалонил-КоА в сукцинил-КоА (см. рис. 4). Ферменту метилмалонил-КоА-мутазе необходим аденозилкобаламин (метаболит витамина В12) в качестве кофермента. Дефицит мутазы/ ее кофермента приводит к накоплению метилмалоновой кислоты и ее предшественников в физиол. жидкостях. Выявлены две биохим. формы дефицита метилмалонил-КоА-мутазы. Их обозначают как mut0 (отсутствие детектируемой активности фермента) и mut- (остаточная, но недостаточная активность мутазы). Пациенты с метилмалоновой ацидемией, обусловленной дефицитом апофермента мутазы (mut0), не отвечают на терапию гидроксокобаламином.
У остальных пациентов с метилмалоновой ацидемией нарушение обусловлено образованием аденозилкобаламина из витамина В12, поступающего с пищей. Для абсорбции витамина В12 из пищевых источников в терминальном отделе подвздошной кишки требуется внутренний фактор (гликопротеин, секретируемый париетальными клетками желудка). Его переносчиками в крови служат гаптокоррин и транскобаламин II. Комплекс транскобаламина и II-кобаламина (ТСII-Cbl) распознается специфическим рецептором на клеточной мембране (рецептором транскобаламина, обозначаемым CD320) и проникает в клетку посредством эндоцитоза. В лизосоме ТСП-СЫ гидролизуется, и при участии LMBRD1 (cblF) и ABCD4 (cblJ) свободный cbl высвобождается в цитозоль (см. рис. 4). Мутации генов LMBRD1/ABCD4 нарушают высвобождение cbl из лизосом. В цитоплазме cbl связывается с белком ММАСНС (см. далее cblC), который удаляет фрагмент, присоединенный к кобальту в молекуле сЫ, и восстанавливает степень окисления кобальта от +3 (коб (III) аламин) до +2 (коб (II) аламин).
Затем он попадает в митохондрии, где катализируется посредством ММАВ (cblB) и ММАА (cblA) с образованием аденозилкобаламина, кофермента метилмалонил-КоА-мутазы. Согласно др. направлению пути, цитозольный cbl и метионинсинтазоредуктаза (cblE) образуют метилкоба-ламин, действующий как кофермент для метионинсинтазы (cblG, см. рис. 3). Белок MMADHC (см. cblD), по-видимому, участвует в определении пути cbl (входит ли он в митохондрии/остается в цитоплазме).
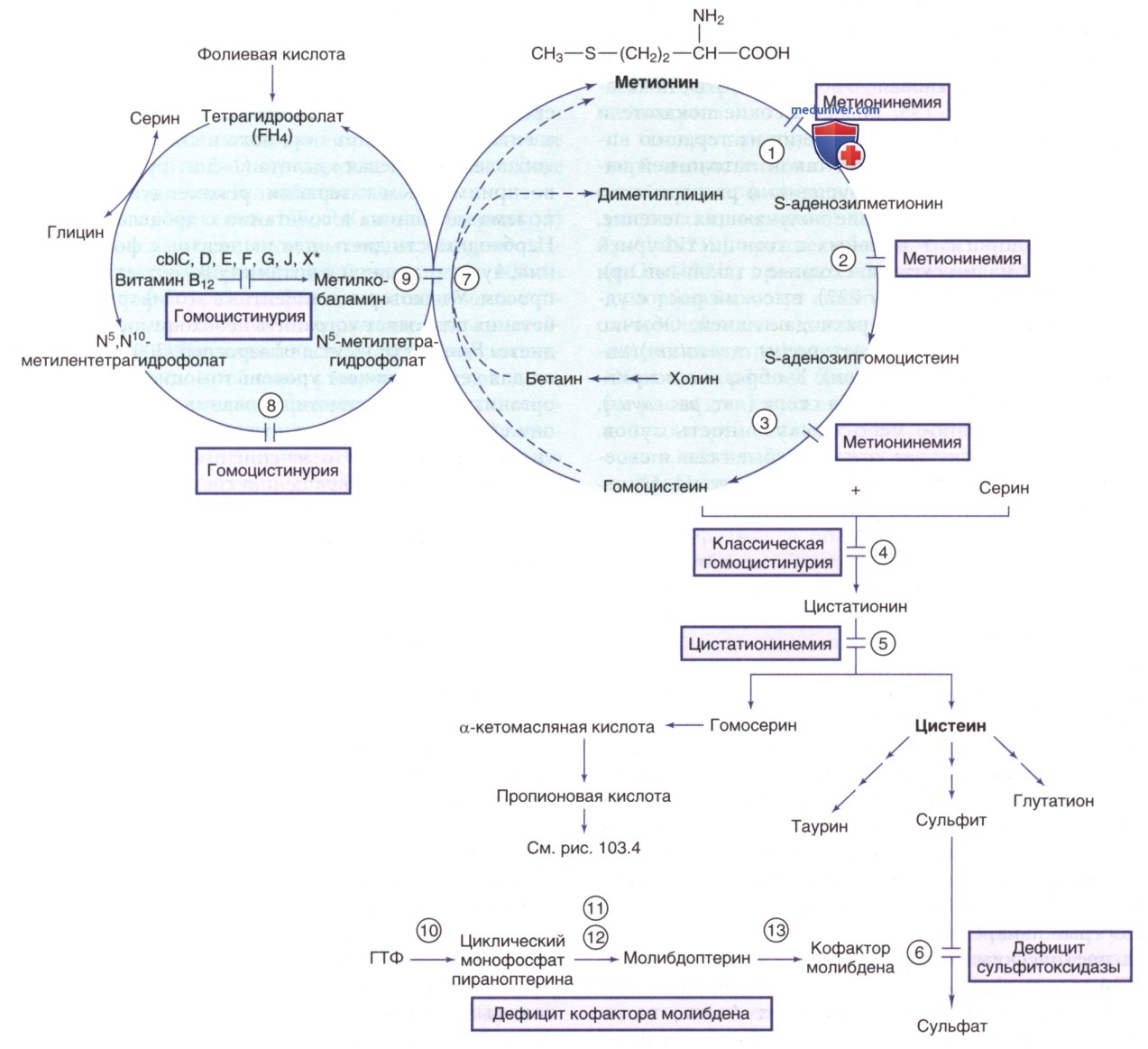
Рисунок 3. Пути метаболизма серосодержащих аминокислот. Ферменты: 1) метионинаденозилтрансфераза (MATI/III); 2) глицин-N-метилтрансфераза, 3) 5-аденозилгомоцистеингидролаза; 4) цистатионинсинтаза; 5) цистатионаза; 6) сульфитоксидаза; 7) бетаингомоцистеинметилтрансфераза; 8) метилентетрагидрофолатредуктаза; 9) метионинсинтаза (cbIG); 10) белок 1 биосинтеза кофактора молибдена; 11) молибдоптеринсинтаза; 12) аденилилтрансфераза и серотрансфераза (MOCS3); 13) гефирин. *Нарушения в кобаламине (cbl) С, D, F, J, X приводят к метилмалоновой ацидемии и гомоцистинурии
У людей с патогенными вариантами, влияющими на рецептор транскобаламина (CD320), расположенный на поверхности клетки, нарушено поглощение ТСII-Cbl клетками. У лиц, гомозиготных по патогенным вариантам гена CD320, кодирующего рецептор транскобаламина, м.б. небольшое повышение содержания метилмалоновой кислоты в крови и моче. Данных пациентов можно выявить при скрининге новорожденных на основании повышенного содержания пропионилкарнитина (С3). При дефиците рецепторов транскобаламина содержание метилмалоновой кислоты и пропионилкарнитина в плазме обычно нормализуется в 1-й год жизни. Неясно, существует ли долгосрочный клинический фенотип, обусловленный этим нарушением.
Выявлено девять разл. дефектов в/клеточного метаболизма cbl. Они обозначаются cblA-cblG, cblJ и cblХ, где cbl означает дефект на любой стадии метаболизма сЫ. Дефекты cblA, сЫВ и cblD вариант 2 вызывают только метилмалоновую ацидемию. У пациентов с дефектами cblС, классическим cblD, cblD, cblJ и cblX нарушается синтез как аденозилкобаламина, так и метилкобаламина, в результате чего развивается комбинация метилмалоновой ацидемии и гомоцистинурии. Дефекты cblD вариант 1, cblE и cblG влияют только на синтез метилкобаламина, что приводит к развитию гомоцистинурии без метилмалоновой ацидурии.
Биохим. проявления у пациентов с изолированной метилмалоновой ацидемией, вызванной mut0, mut-, cblА, cblB и cblD вариант 2, перекрываются. Различия в тяжести клинического течения варьируют от тяжелобольных новорожденных до взрослых, кажущихся здоровыми. При тяжелых формах в первые несколько дней жизни могут развиться заторможенность, проблемы со вскармливанием, рвота, сепсисоподобная картина, тахипноэ (вследствие метаболического кетоацидоза) и гипотония, которые при отсутствии лечения могут прогрессировать до гипераммониемической энцефалопатии, комы и летального исхода. У младенцев, переживших первый приступ, могут развиваться аналогичные острые метаболические эпизоды при повышенном катаболизме, напр., спровоцированном инфекциями, длительным голоданием и приемом пищи с высоким содержанием белка. В определенных ситуациях такие острые нарушения могут приводить к внезапному повреждению базальных ганглиев (центров движения в ЦНС), метаболическому инсульту, в результате чего развивается изнурительное двигательное расстройство.
В перерывах между острыми приступами у пациента, как правило, сохраняется гипотония и проблемы с вскармливанием. С возрастом возникают др. осложнения заболевания, в т.ч. рецидивирующий панкреатит, угнетение функции костного мозга, остеопения и атрофия зрительного нерва. Среди взрослых пациентов зарегистрированы случаи ХПН и тубулоинтерстициального нефрита, требующего трансплантации почки. Более тяжелые почечные осложнения развиваются у пациентов с mut0 и тяжелой сЫВ формами метилмалоновой ацидемии. У пациентов с легкими формами с возрастом могут появиться гипотония, задержка физического и психомоторного развития. Умственное развитие пациентов с легкой метилмалоновой ацидемией может оставаться в пределах нормы.
Эпизодический характер заболевания и его биохим. изменения у некоторых пациентов можно спутать с таковыми при приеме этиленгликоля внутрь (антифриза). Пик пропионата в образце крови младенца с метилмалоновой ацидемией был ошибочно принят за этиленгликоль при анализе методом газовой хроматографии без масс-спектрометрии.
К лабораторным признакам относятся кетоз, метаболический ацидоз, гиперглицинемия, гипераммониемия, гипогликемия, анемия, нейтропения, тромбоцитопения и наличие большого количества метилмалоновой кислоты в физиол. жидкостях (см. рис. 6). В моче также присутствуют метаболиты пропионовой кислоты (3-гидроксипропионат и метилцитрат). Профиль ацилкарнитина в плазме выявляет повышенный уровень пропионилкарнитина (С3) и метилмалонилкарнитина (C4DC). Гипераммониемию при метилмалоновой ацидемии можно спутать с нарушением цикла мочевины. Однако пациенты с дефектами ферментов цикла мочевины обычно не страдают от ацидоза, при этом обнаруживается высокий уровень глутамина в плазме крови (рис. 12). Причина гипераммониемии не совсем понятна, но, вероятно, связана с ингибированием проксимального цикла мочевины в митохондриальном матриксе.
Рисунок 12. Цикл мочевины: пути экскреции аммиака и метаболизма орнитина. Реакции, протекающие в митохондриях, показаны фиолетовым цветом. Реакции, показанные пунктирными стрелками, представляют собой альтернативные пути экскреции аммиака. Ферменты: 1) карбамиолфосфатсинтетаза типа 1 (КФС1); 2) орнитинтранскарбамилаза (ОТК); 3) аргининосукцинатсинтетаза (АСС); 4) аргининосукцинатлиаза (АСЛ); 5) аргиназа 1 ; 6) орнитинаминотрансфераза; 7) N-ацетилглутамат (НАГ) синтетаза; 8) цитрин; 9) транспортер орнитина (0РНТ1). Синдром ГГГ — синдром гиперорнитинемии-гипераммониемии-гомоцитруллинемии
Диагноз устанавливается на основании выявления мутаций в причиннозначимом гене в ходе измерения включений пропионата методом комплементации в культуре фибробластов и измерения удельной активности фермента мутазы в биоптатах/клеточных экстрактах.
Острые приступы лечат так же, как и пропионовую ацидемию. Длительное лечение заключается в соблюдении низкобелковой диеты с рекомендуемой нормой потребления L-карнитина (внутрь 50-100 мг/кг в сутки). Пациентам с тяжелыми формами метилмалоновой ацидемии могут потребоваться изменения белковой диеты, аналогичные тем, которые назначают пациентам с пропионовой ацидемией. Пациенты с изолированной метилмалоновой ацидемией, вызванной дефектами в/клеточного метаболизма cbl (cblА, cblD вариант 2 и некоторые пациенты с cblВ), отвечают на парентеральное введение гидроксокобаламина. Для коррекции хронического ацидоза обычно требуется хроническая заместительная терапия бикарбонатом. В перерывах между приступами уровень аммиака в плазме обычно нормализуется, и хроническое лечение гипераммониемии требуется редко. Стрессовые ситуации, которые могут спровоцировать острые приступы (инфекция, длительное голодание, травмы, операции, высокобелковая пища), следует купировать незамедлительно.
Частыми осложнениями при длительном ведении таких пациентов являются недостаточное питание вследствие плохого аппетита, чрезмерное ограничение белка и дефицит незаменимых аминокислот. Поэтому в начале терапии часто рекомендуется энтеральное питание через гастростому. Для обеспечения питания, соответствующего метаболическим потребностям пациента, необходим тщательный мониторинг pH крови, содержания незаменимых аминокислот, концентрации метилмалоната в крови и моче и параметров физического развития. Кроме того, для раннего выявления и лечения хронических осложнений необходим частый мониторинг функции почек, зрения, слуха и минеральной плотности костей. Описан дефицит глутатиона, поддающийся лечению аскорбатом.
У возрастающего числа пациентов предпринимаются попытки трансплантации печени, почек и комбинированной трансплантации почек и печени. Трансплантация печени/печени и почки может облегчить, но не устранить метаболические нарушения. Трансплантаты печени/печени и почки не обеспечивают полной защиты от метаболического инсульта. Сама по себе трансплантация почки может восстановить функцию почек, но приводит лишь к незначительному улучшению и клинической стабилизации пациентов.
Прогноз зависит от выраженности симптомов и развития осложнений. В целом пациенты с полным отсутствием апофермента мутазы (mut0) и тяжелыми формами дефицита cblВ имеют наименее благоприятный прогноз, а пациенты с дефектами mut- и cblA — хороший.
Метилмалоновая ацидемия м.б. выявлена при скрининге новорожденных путем измерения пропионилкарнитина (С3) методом тандемной масс-спектрометрии. По оценкам, частота встречаемости всех форм метилмалоновой ацидурии составляет 1:50 000-100 000 живорождений. Все дефекты, вызывающие изолированную метилмалоновую ацидемию, наследуются по АуР-типу. Ген мутазы (MUT) расположен на коротком плече хромосомы 6р12.3. Зарегистрированы случаи новорожденных с метилмалоновой ацидемией и тяжелым СД, вызванным агенезом β-клеток, у которых имеется изодисомия хромосомы 6 со стороны отца. У пациентов с данной патологией были выявлены мутации генов МА (ММАА в хромосоме 4q31.21), МВ (ММАВ в хромосоме 12q24.11) и всех форм cblD (MMADHC в хромосоме 2q23.2). Вышеуказанная группа cblH идентична cblD вариант 2.
р) Комбинированная метилмалоновая ацидурия и гомоцистинурия (дефекты кобаламинов C, D, F, J и X). Комбинированная метилмалоновая ацидемия и гомоцистинурия, вызванная дефицитом CblC, является наиболее распространенным типом в/клеточных нарушений биосинтеза cbl (витамина В12). Дефицит cblC встречается так же часто, как и дефицит метилмалонил-КоА-мутазы. Др. нарушения (cblD, cblF, cblJ, MX) встречаются гораздо реже (см. рис. 3, 4). У пациентов с дефектами cblС, cblD и cblX наблюдаются неврологические симптомы. У большинства пациентов дефект cblС проявляется в 1-й месяц жизни в виде задержки физического и психомоторного развития, вялости, нарушения вскармливания, нистагма и судорог. В отличие от изолированной метилмалоновой ацидемии mut-типа, гипераммониемия наблюдается нечасто, а гиперглицинемия отсутствует. ЗВУР и микроцефалия позволяют предположить, что у некоторых детей cblС может проявляться пренатально. Зарегистрированы случаи пациентов с поздним началом заболевания и внезапным развитием деменции и миелопатии, даже во взрослом возрасте. У пациентов с дефектом cblС часто наблюдается мегалобластная анемия.
В крови обнаруживается легкое/умеренное повышение концентрации метилмалоновой кислоты и значительное повышение общего гомоцистеина плазмы. В отличие от классической гомоцистинурии, у пациентов с cblC при отсутствии лечения наблюдается низкое/нормальное содержание метионина в плазме. Часто развиваются патологии сетчатки (напр., макулопатия в виде бычьего глаза), приводящие к тяжелой прогрессирующей потере зрения. Эти осложнения могут наблюдаться уже в 3 мес даже у выявленных пациентов, получающих требуемое лечение. Может развиться тромботическая микроангиопатия, проявляющаяся в виде ГУС, легочной гипертензии и легочного сердца. У пациентов с дефектом сЫС зарегистрированы такие осложнения, как гидроцефалия и некомпактная кардиомиопатия.
Подобно пациентам с МС, у мальчиков с cblX наблюдается повышение общего гомоцистеина и метилмалоновой кислоты в плазме, но повышение данных метаболитов обычно умеренное. В отличие от пациентов с дефицитом cblC, которые обычно поддаются лечению, у пациентов с дефицитом cblX наблюдаются задержка физического развития, серьезная задержка психического развития и трудноизлечимая эпилепсия, несмотря на интенсивное лечение.
Клинические проявления при дефиците cblF весьма разнообразны. У пациентов в 1-й месяц жизни могут наблюдаться проблемы с вскармливанием, задержка физического и психического развития и стойкий стоматит. При поздней диагностике и несвоевременном начале терапии могут наблюдаться гиперпигментация кожи, задержка психомоторного развития, умственная отсталость и низкий рост. У пациентов с дефектом cblF наблюдались мальабсорбция витамина В12 и низкий его уровень в плазме. Клинические проявления дефекта cblJ в большой степени совпадают с симптомами при дефиците cblF. У некоторых пациентов с дефектами cblF и cblJ зарегистрированы случаи дисморфизма и ВПС.
Имеется ограниченный опыт лечения пациентов с дефектами cblC, cblD, cblF, cblJ и cblX. Большие дозы [гидроксокобаламина (до 0,3 мг/кг в сутки) + бетаина (до 250 мг/кг в сутки)] дают биохим. улучшение с переменным клиническим эффектом. Пациенты с дефицитом cblF и cblJ обычно показывают «+» биохим. и клинический ответ на меньшие дозы гидроксокобаламина (парентерально, от 1 мг/нед до 1 мг/сут). Рекомендуется прием фолиевой/фолиниевой кислот. Следует избегать дефицита метионина в питании.
Причиной нарушения cblC являются мутации гена ММАСНС (в хромосоме 1р34.1). В 40% аллелей ММАСНС наблюдается вариант со сдвигом рамки (c.271dupA), который обусловливает менее благоприятный клинический исход. Причиной нарушения cb/D являются мутации гена MMADHC (в хромосоме 2q23.2). Мутации, приводящие к варианту 1 cb/D (вызывающему только гомоцистинурию), влияют на С-концевой домен гена; приводящие к варианту 2 cblD (вызывающему только метилмалоновую ацидурию) влияют на N-концевой домен. Пациенты с классической cblD, как с гомоцистинурией, так и с метилмалоновой ацидемией, имеют мутации, приводящие к снижению экспрессии белка. Причиной нарушения cblF являются мутации гена LMBRD1 (в хромосоме 6q13), кодирующего белок лизосомальной мембраны. Нарушение cblJ вызвано мутациями в гене ABCD4 (в хромосоме 14q24.3), кодирующем АТФ-связывающий кассетный белок, расположенный на лизосомной мембране. Причиной нарушения cblX являются мутации гена HCFC1 в Х-хромосоме (Xq28), который кодирует фактор транскрипции, вероятно, необходимый для экспрессии гена ММАСНС. Это единственное Х-сцепленное нарушение во в/клеточном метаболизме В12.
с) Изолированная гомоцистинурия. У пациентов с cblD вариант 1, cblE и cblG наблюдается изолированная гомоцистинурия без метилмалоновой ацидемии (см. «Гомоцистинурия, вызванная нарушением синтеза метилкобаламина» - просим Вас пользоваться формой поиска по сайту выше).
т) Комбинированная малоновая и метилмалоновая ацидурия (ACSF3-ассоциированное нарушение). Комбинированная малоновая и метилмалоновая ацидурия представляет собой редкое АуР-заболевание, возникающее в результате мутаций ACSF3. ACSF3 — это предположительно ацил-КоА-синтетаза, необходимая для превращения малоновой и метилмалоновой кислот в их КоА-производные в митохондриальном матриксе. Нарушение можно заподозрить при повышенном содержании малоновой и метилмалоновой кислот в моче и плазме. Оно отличается от дефицита малонил-КоА-декарбоксилазы тем, что содержание метилмалоновой кислоты в моче превышает концентрацию малоновой в 5 раз. Пропионилкарнитин (СЗ-карнитин) в плазме пациентов с комбинированной малоновой и метилмалоновой ацидурией соответствует норме, поэтому универсальные программы скрининга новорожденных с использованием СЗ-карнитина в пятнах крови для выявления метилмалоновой ацидемии не обнаруживают это заболевание. Клинический фенотип изучен не до конца.
Сообщалось, что пациенты молодого возраста, проспективно выявленные в младенчестве в рамках скрининга новорожденных, были бессимптомны, но отдаленные результаты в данной группе требуют дальнейшего изучения. У взрослых пациентов, выявленных клинически, наблюдаются разл. симптомы, в т.ч. метаболические кризисы, задержка физического развития, судороги, проблемы с памятью, атрофия зрительного нерва/спинного мозга и прогрессирующая нейродегенерация. Лечение данной патологии поддерживающее и включает отказ от питания с чрезмерным содержанием белка. Прием витамина В12, по всей видимости, не снижает концентрации метаболитов малоновой и метилмалоновой кислоты в физиол. жидкостях.