Фенилаланин (ФА) является незаменимой аминокислотой. ФА, поступивший с пищей и не использованный для синтеза белка, обычно расщепляется тирозиновым путем (рис. 1).
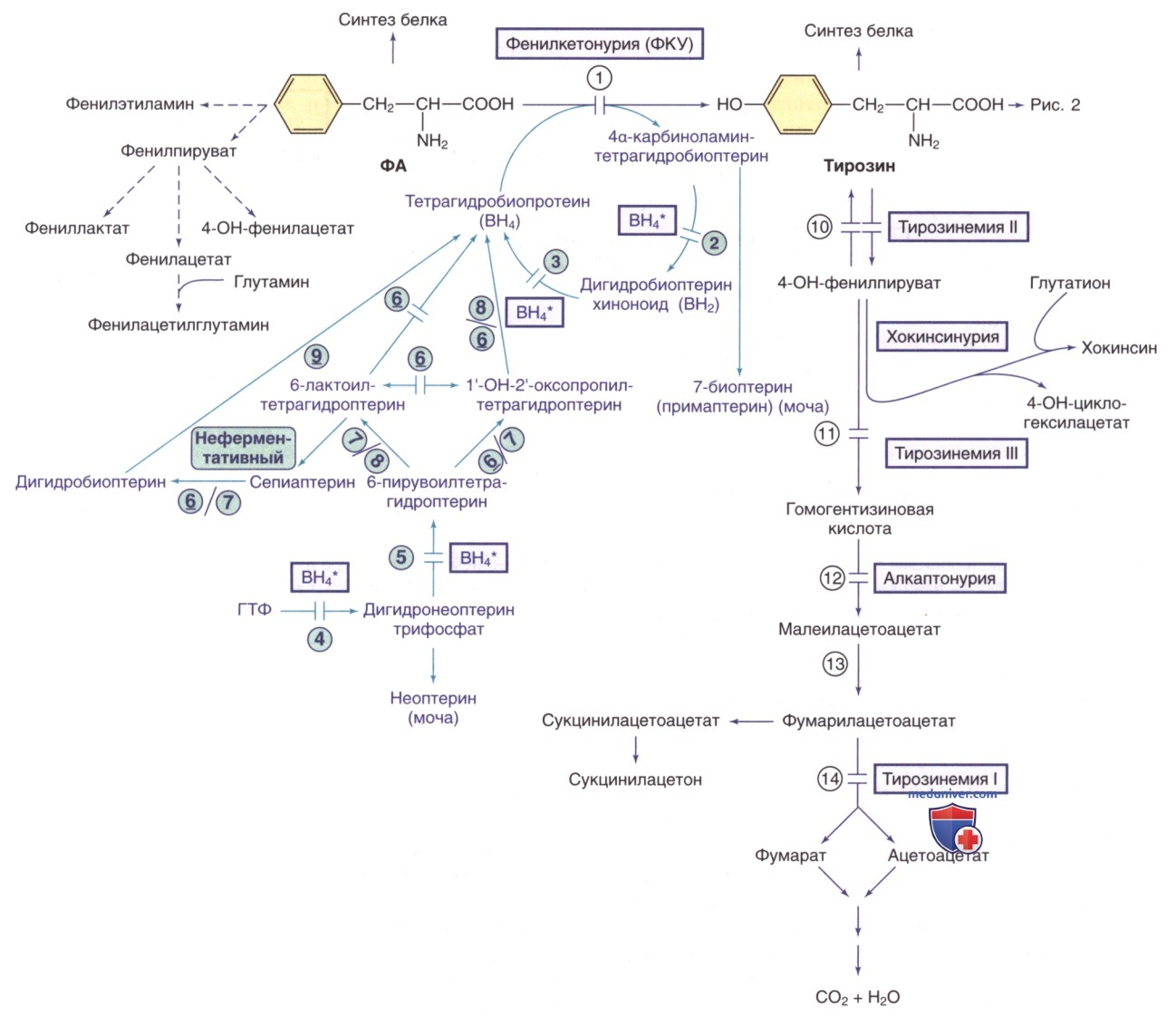
Рисунок 1. Пути метаболизма фенилаланина (ФА) и тирозина. Генетически детерминированные ферментативные нарушения показаны горизонтальными линиями, пересекающими стрелку(-и) реакции. Пути синтеза кофактора тетрагидробиоптерина (ВН4) показаны фиолетовым цветом. ВН4* относится к дефектам метаболизма ВН4, влияющим на гидроксилазы ФА, тирозина и триптофана (рис. 2 и 5). Ферменты: 1) фенилаланингидроксилаза; 2) птеринкарбиноламиндегидратаза; 3) дигидробиоптеринредуктаза; 4) гуанозинтрифосфат (ГТФ)-циклогидролаза; 5) 6-пирувоилтетрагидроптеринсинтаза; 6) сепиаптеринредуктаза; 7) карбонилредуктаза; 8) альдолазоредуктаза; 9) дигидрофолатредуктаза; 10) тирозинаминотрансфераза; 11) 4-гидроксифенилпируватдиоксигеназа; 12) диоксигеназа гомогентизиновой кислоты; 13) малеилацетоацетат изомераза; 14) фумарилацетоацетат гидролаза
Для синтеза белка используется небольшая часть ФА, его основное количество утилизируется путем превращения в тирозин, являющегося субстратом для синтеза биогенных аминов и меланина. Превращение L-фенилаланина в L-тирозин осуществляется с помощью фермента фенилаланингидроксилазы (ФАГ). Активность фермента ФАГ зависит от трех основных кофакторов: ФАГ-стимулирующего белка (ФАГС), тетрагидробиоптерина (BH4) и молекулярного кислорода. Функция BH4 заключается в стабилизации четвертичной структуры (фолдинге) ФАГ и др. ферментов, участвующих в гидроксилировании тирозина, триптофана, аргинина*.
P.S. *КР «Классическая фенилкетонурия и другие виды гиперфенилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.
Дефицит фермента ФАГ/его кофактора BH4 вызывает накопление ФА в жидких средах организма и ГМ.
Повышение уровня ФА в плазме зависит от степени дефицита фермента. У пациентов с тяжелым дефицитом ФАГ (прежнее название — классическая ФКУ) значения ФА в плазме без специальной диеты обычно >20 мг/дл (>1200 мкмоль/л). У пациентов с умеренными патологическими вариантами ФАГ содержание ФА в плазме составляет 10-20 мг/дл (600-1200 мкмоль/л). У пациентов с легкой гиперфенилаланинемией (ГФА) без специальной диеты содержание ФА в плазме 2-10 мг/дл (120— 600 мкмоль/л). У больных детей грудного возраста с такой патологией при концентрации ФА в плазме >20 мг/дл его избыток метаболизируется до фенилкетонов (фенилпируват и фенилацетат; см. рис. 1), которые выделяются с мочой, что позволяет обоснованно применять при данном нарушении метаболизма термин ФКУ
Роль этих метаболитов в патогенезе поражения ЦНС у пациентов с ФКУ неизвестна; их присутствие в жидких средах организма определяет тяжесть состояния. ГМ является основным органом, поражаемым при ФКУ, но точный механизм повреждения остается невыясненным. Могут иметь значение и повышенное содержание ФА до токсичного уровня, и недостаточное количество тирозина. ФАГ преобразует ФА в тирозин, требуемый для выработки нейромедиаторов, таких как адреналин, норадреналин и дофамин (рис. 2). При тяжелой степени ферментативного блока тирозин становится незаменимой аминокислотой, поэтому при недостаточном поступлении с пищей может возникнуть его дефицит.
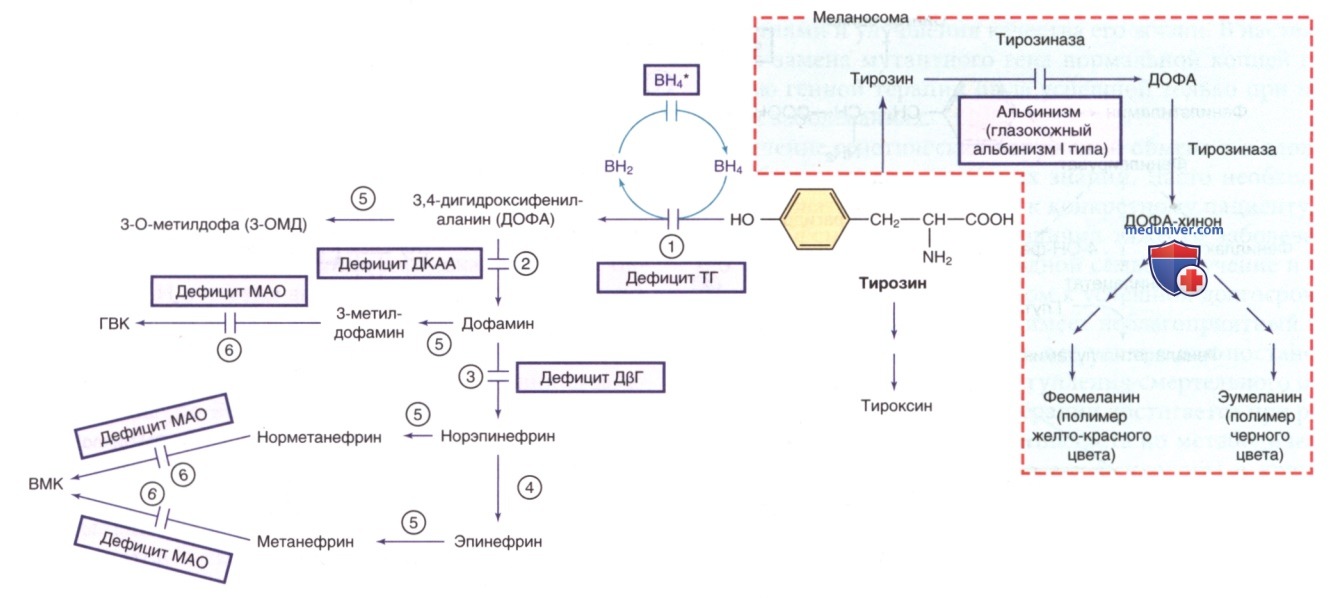
Рисунок 2. Другие пути метаболизма тирозина. ВН4* указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). ГВК — го-мованиловая кислота; ВМК — ванилилминдальная кислота; ВН4 — тетрагидробиоптерин. Ферменты: 1) тирозингидроксилаза (ТГ); 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) дофамин-β-гидроксилаза (ДβГ); 4) фенилэтаноламин-N-метилтрансфераза (ФНМТ); 5) катехол-О-метилтрансфераза (КОМТ); 6) МАО
С др. стороны, при более низкой концентрации ФА в плазме и ткани ГМ наблюдается улучшение нейроповеденческих показателей, это подтверждает мнение о том, что токсические уровни ФА играют ключевую роль в механизмах развития заболевания. Высокий уровень ФА в крови может насыщать транспортную систему через ГЭБ и вызывать ингибирование церебрального поглощения др. крупных нейтральных аминокислот, таких как аминокислоты с разветвленной цепью, тирозин и триптофан, при этом нарушая синтез белка в ГМ.
На основании уровня ФА в крови до лечения выделяют легкую форму ГФА (120-360 мкмоль/л/2-6 мг/дл), умеренную (360-1200 мкмоль/л/6-20 мг/дл), тяжелую (>1200 мкмоль/л/>20 мг/дл)*.
P.S. КР «Классическая фенилкетонурия и другие виды гиперфенилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.
а) Тяжелая степень дефицита фенилаланингидрокислазы (классическая фенилкетонурия). Повышение уровня ФА в плазме >20 мг/дл (>1200 мкмоль/л) при отсутствии лечения неизменно приводит к развитию признаков и симптомов классической ФКУ, за исключением редких и непредсказуемых случаев.
1. Клинические проявления. Новорожденный с такой патологией при рождении выглядит нормальным. Если ребенок остается без лечения, то постепенно развивается глубокая умственная отсталость. В течение первых нескольких месяцев задержка развития когнитивных функций может не проявляться. При этом у 50-70% пациентов, не получающих лечение, IQ будет <35, а у 88-90% — <65. Только 2-5% пациентов без лечения могут обладать нормальными умственными способностями. Ранним симптомом болезни м.б. рвота, порой достаточно сильная, из-за чего иногда ошибочно диагностируют пилоростеноз. У детей старшего возраста, не получавших лечение, проявляется СДВГ с РАС, в т.ч. бесцельные движения руками, ритмичное покачивание и атетоз.
В грудном возрасте дети, не получающие/получающие недостаточное лечение, имеют более светлый цвет кожи, чем их здоровые братья и сестры. У некоторых может наблюдаться себорейная/экзематозная сыпь, которая обычно проявляется в легкой форме и исчезает по мере взросления ребенка. У таких детей присутствует запах фенилуксусной кислоты, который описывают как затхлый/«мышиный». Из неврологических признаков появляются судороги (25%), спастичность, гиперрефлексия и тремор; >50% имеют изменения на ЭЭГ. Др. частыми симптомами у детей, не получающих лечение при этой патологии, являются микроцефалия, выступающие верхние челюсти с широко расставленными зубами, гипоплазия эмали и задержка роста. У пациентов всех возрастов с ФКУ зарегистрирована низкая минеральная плотность костей и остеопения.
Хотя основной причиной считается недостаточное потребление природных белков, точный патогенез этого осложнения остается неясным. Наиболее эффективное долгосрочное лечение ФКУ достигается при работе группы опытных специалистов регионального мед. центра (специалиста по метаболизму, диетолога и психолога). В странах, где действуют программы неонатального скрининга ФКУ, ее клинические проявления наблюдаются редко.
- Гиперфенилаланинемия, не сопровождающаяся фенилкетонурией. В любой программе скрининга на ФКУ определяется группа новорожденных, у которых исходные концентрации ФА в плазме выше нормы (>2 мг/дл/120 мкмоль/л), но <20 мг/дл (1200 мкмоль/л). У таких детей фенилкетоны обычно не выделяются. Пациентам с ГФА, не сопровождающейся ФКУ, при отсутствии лечения в зависимости от содержания ФА в плазме может потребоваться диета. Предпринимались попытки разделить пациентов на разл. подгруппы в зависимости от степени ГФА, но такая практика имеет мало клинических/терапевтических преимуществ. Все дети с более легкими формами ГФА должны быть обследованы на потенциальный дефицит BH4.
2. Диагностика. Поскольку ранние клинические симптомы (рвота, задержка развития и экзематозная сыпь) развиваются постепенно и не являются специфическими, во всех развитых странах диагностику ГФА обычно включают в программу скрининга новорожденных. У детей с «+» результатами скрининга диагноз должен подтверждаться количественным измерением концентрации ФА в плазме. Выявление и измерение фенилкетонов в моче не включено ни в одну программу скрининга. В странах и регионах, где такие программы не действуют, простым тестом для обследования грудных детей с нарушениями развития и неврологическими отклонениями может стать определение фенилкетонов в моче с использованием хлорида железа.
После диагностики ГФА необходимо провести дополнительные исследования метаболизма ВН4 для исключения его дефицита как причины ГФА.
У женщин репродуктивного возраста рекомендуется исключение ГФА, если в анамнезе есть данные о невынашивании беременности/рождение детей с ВПР*.
P.S. *КР «Классическая фенилкетонурия и другие виды гиперфе-нилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.
- Неонатальный скрининг на гиперфенилаланинемию. В США и многих др. странах используются эффективные и относительно недорогие методы массового скрининга новорожденных. Для анализа используют несколько капель крови, которые помещают на фильтровальную бумагу и отправляют в центральную лабораторию. Как метод скрининга применяется тандемная масс-спектрометрия, которая идентифицирует все формы ГФА с низкой частотой л/п результатов, высокой точностью и достоверностью получаемых данных. Добавление молярного соотношения ФА/тирозин дополнительно снизило число л/п результатов.
Диагноз должен быть подтвержден измерением концентрации ФА в плазме. ФА в крови у детей с ФКУ может повышаться до диагностических уровней уже через 4 ч после рождения, даже при отсутствии белкового питания. Рекомендуется брать кровь для скрининга в первые 24-48 ч жизни после белкового питания, поскольку это позволяет снизить вероятность получения л/о результатов, особенно при легких формах заболевания.
Главным ДК ГФА считается повышенное содержание ФА в крови >2,0 мг/дл (120 мкмоль/л), при «+» результате проводится ретест. Верификация диагноза ГФА проводится с помощью молекулярно-генетического исследования и определения соотношения ФА/тирозин, которое в норме <1, соотношение >3 свидетельствует о высокой вероятности наличия у больного недостаточности ФАГ*.
P.S. *КР «Классическая фенилкетонурия и другие виды гиперфе-нилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.
3. Лечение. Основой лечения ФКУ является диета с низким содержанием ФА. Согласно общему мнению специалистов, если у пациента уровень ФА в крови >10 мг/дл (600 мкмоль/л), следует немедленно начинать диетотерапию. Считается, что детям грудного возраста со стойкими (более нескольких дней) уровнями ФА в плазме >6 мг/дл (360 мкмоль/л) также следует назначать диету с ограничением ФА, аналогичную диете при классической ФКУ. Терапия направлена на снижение уровня ФА в плазме и ГМ.
В продаже имеются специализированные молочные формулы без ФА/с его низким содержанием. Необходимо начинать диету сразу после установления диагноза. Поскольку ФА не синтезируется эндогенно, его содержание в рационе должно предотвращать развитие его дефицита. Переносимость ФА с пищей определяется в зависимости от возраста и тяжести дефицита ФАГ.
При дефиците ФА наблюдаются летаргия, задержка физического развития, анорексия, анемия, сыпь, диарея, также возможен летальный исход. Кроме того, при таком нарушении обмена тирозин становится незаменимой аминокислотой и должен поступать в организм в достаточном количестве. В продаже имеются специальные продукты питания с низким содержанием ФА для диетического лечения детей и взрослых.
Лечение в периоде новорожденности следует начинать до достижения возраста 10 дней. Задержка начала лечения на каждые 4 нед приводит к снижению показателя IQ на 4 балла, поэтому целесообразно начинать лечение не позднее 3 нед жизни. Не рекомендуется назначать лечение ранее нелеченым пациентам с концентрацией ФА крови <360 мкмоль/л. В этом случае необходимо осуществлять регулярный контроль ФА с целью своевременного назначения терапии.
Ни мед. центры в США, ни специалисты др. стран не достигли однозначного консенсуса относительно оптимальных уровней ФА в крови у пациентов с этим заболеванием. В настоящее время рекомендуется поддерживать уровень ФА в крови 2-6 мг/дл (120-360 мкмоль/л) на протяжении всей жизни. Прекращение терапии даже во взрослом возрасте может привести к снижению IQ и когнитивных функций. Однако очень трудно соблюдать диету с низким содержанием ФА на протяжении всей жизни. Пациенты, придерживающиеся диеты с ограничением ФА в детстве, но прекращающие ее в подростковом/взрослом возрасте, могут испытывать значительные трудности с исполнительными функциями, концентрацией, эмоциональностью и депрессией. Нарушение исполнительных функций (когнитивная способность контролировать и регулировать поведение) также может возникать у детей, начавших лечение в раннем возрасте, даже несмотря на соблюдение диеты.
Диетотерапия при сопутствующих заболеваниях (выраженная гипертермия, интоксикация, разл. диспепсические явления) и при отказе от приема аминокислотной смеси включает кратковременное (2-3 сут) прекращение диетотерапии с заменой продуктов лечебного питания на натуральные с невысоким содержанием белка. По окончании острого периода заболевания специализированный лечебный продукт вновь вводится в рацион, только за более короткий период, чем в начале диетотерапии.
Поскольку соблюдать строгую диету с низким содержанием ФА сложно, продолжаются попытки найти др. методы лечения таких пациентов. Еще один подход к диетической терапии заключается в введении больших нейтральных аминокислот. Они (тирозин, триптофан, лейцин, изолейцин, валин, метионин, гистидин и ФА) имеют один и тот же транспортный белок (типа 1/LAT-1) для прохождения через мембрану кишечных клеток и ГЭБ. Связывание больших нейтральных аминокислот с транспортным белком является конкурентным процессом. Применение этих аминокислот обосновывается тем, что указанные молекулы конкурируют с ФА за связь с общим транспортером через ГЭБ; следовательно, большие концентрации др. больших нейтральных аминокислот в просвете кишечника и крови снижают поступление ФА в кровоток и ГМ.
Для определения эффективности такого лечения необходимо проведение крупных контролируемых клинических исследований.
Пероральное введение ВН4, кофактора ФАГ, может привести к снижению уровня ФА в плазме у некоторых пациентов с дефицитом ФАГ. Уровни ФА в плазме у этих пациентов могут снизиться настолько, что это позволит существенно изменить диетические ограничения. В очень редких случаях диету можно прервать, если уровень ФА остается <6 мг/дл (360 мкмоль/л). Невозможно однозначно предсказать ответ на терапию ВН4, основываясь только на генотипе, особенно у пациентов со сложными гетерозиготными состояниями. Сапроптерина дигидрохлорид (синтетическая форма ВН4), действующий как кофактор у пациентов с остаточной активностью ФАГ, утвержден FDA для снижения уровня ФА при ФКУ. Устойчивое снижение уровня ФА в плазме на <30% соответствует ответу на введение сапроптерина. Пегилированная рекомбинантная фенилаланинаммнонийлиаза для инъекций находится в стадии разработки.
Всем пациентам с ГФА рекомендуются профилактические дозы витамина D для профилактики остеопении. В случае необходимости проводится сопутствующая посиндромная терапия. В РФ функционируют школы ФКУ для обучения пациентов/родителей/законных представителей управлению заболеванием/самоконтролю*.
P.S. * КР «Классическая фенилкетонурия и другие виды гиперфенилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.
- Беременность у женщин с дефицитом фенилаланингидроксилазы (материнская фенилкетонурия). Беременные женщины с дефицитом ФАГ, не соблюдающие диету с ограничением ФА, имеют очень высокий риск рождения ребенка с умственной отсталостью, микроцефалией, задержкой роста, ВПР и ВПС. Эти осложнения напрямую связаны с повышенным уровнем ФА в крови матери во время беременности. Будущие матери, соблюдавшие рекомендованную диету в связи с дефицитом ФАГ, должны ее придерживаться до и во время беременности. Имеются данные, что наилучшие результаты достигаются при строгом контроле концентрации ФА в крови женщины еще до беременности. Содержание ФА в плазме >6 мг/дл (360 мкмоль/л) после зачатия коррелирует с увеличением числа случаев ЗВУР и ВПР, а также с более низким IQ.
Однако имеются убедительные доказательства, что контроль уровней ФА после зачатия приводит к улучшению результатов. Рекомендуемая в настоящее время концентрация ФА на протяжении всей беременности составляет 2-6 мг/дл (120-360 мкмоль/л), хотя некоторые группы экспертов рекомендуют соблюдать содержание ФА в плазме <4 мг/дл (<240 мкмоль/л). Все женщины детородного возраста с дефицитом ФАГ должны быть надлежащим образом проинформированы о риске ВПР у их детей.
При материнской ФКУ проводится скрининговое УЗИ при сроках беременности 11-14 нед, 18-21 нед и 30-34 нед*, а также дополнительное экспертное УЗИ плода в срок 15-16 нед и 25-26 нед беременности**.
P.S. * Приказ М3 РФ от 01.11.2012 № 572н «Об утверждении Порядка оказания медицинской помощи по профилю “Акушерство и гинекология”».
P.S. ** КР «Классическая фенилкетонурия и другие виды гиперфенилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.
б) Гиперфенилаланинемия с дефицитом кофактора тетрагидробиоптерина. У 1-3% детей грудного возраста с ГФА дефект обусловлен одним из ферментов, необходимых для синтеза/реактивации кофактора BH4 (см. рис. 1). Если у этих детей ошибочно диагностирована ФКУ, у них может ухудшаться неврологическое состояние, несмотря на адекватный контроль уровня ФА в плазме. BH4 синтезируется из ГТФ посредством нескольких ферментативных реакций (см. рис. 1). BH4 не только действует как кофактор для ФАГ, но и является кофактором для тирозингидроксилазы и триптофангидроксилазы, которые участвуют в биосинтезе дофамина (см. рис. 2) и серотонина (см. рис. 5) соответственно.
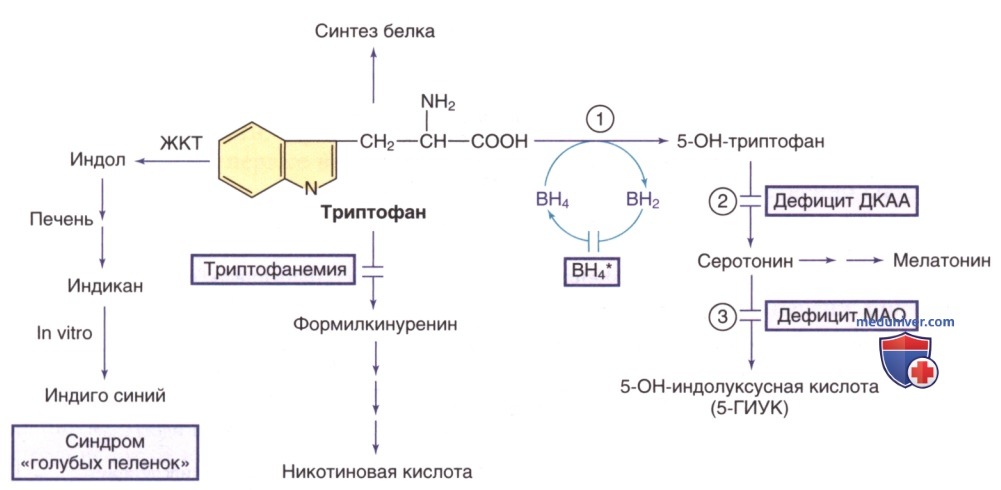
Рисунок 5. Пути метаболизма триптофана. Тетрагидробиоптерин (ВН4*) указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). Ферменты: 1) триптофангидроксилаза; 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) МАО
Следовательно, у пациентов с ГФА, вызванной дефицитом BH4, проявляются неврологические симптомы, обусловленные дефицитом этих нейромедиаторов. Дефицит четырех ферментов, ведущих к дефектному образованию BH4, вызывает ГФА с сопутствующим дефицитом дофамина и серотонина: АуР-дефицит ГТФ-циклогидролазы I, дефицит 6-пирувоилтетрагидроптеринсинтазы, дефицит дигидроптеридинредуктазы и дефицит птерин-4α-карбиноламиндегидратазы. У >1/2 зарегистрированных пациентов наблюдался дефицит 6-пирувоилтетрагидроптеринсинтазы. АуД-формы дефицита ГТФ циклогидролазы I и дефицита сепиаптеринредуктазы приводят к дефициту нейромедиаторов без ГФА.
1. Клинические проявления. Новорожденные с дефицитом кофактора ВН4 выявляются при скрининге на ФКУ по признакам ГФА. Уровни ФА плазмы крови м.б. такими же высокими, как при классической ФКУ/варьировать в пределах более низких величин.
У некоторых пациентов с дефектами гена GCH1 при рождении содержание ФА в крови м.б. в пределах нормы, такие случаи не выявляются при неонатальном скрининге. В дальнейшем у этих пациентов концентрация ФА периодически повышается, в тканях и биологических жидкостях определяется резкое снижение содержания конечных метаболитов биогенных аминов: гомованилиновой и 5-гидроксииндолуксусной кислот.
Кроме того, клинические проявления нарушений метаболизма нейромедиаторов значительно отличаются от таковых при ФКУ. Неврологические симптомы нарушений метаболизма нейромедиаторов часто проявляются в первые несколько месяцев жизни и включают экстрапирамидные признаки (хореоатетоидные/дистонические движения конечностей, центральную и туловищную гипотонию, гипокинезию), затруднения вскармливания и вегетативные расстройства. Также наблюдаются умственная отсталость, судороги, гиперсаливация, затрудненное глотание.
Симптомы обычно прогрессируют и часто имеют выраженные суточные колебания. Прогноз и исход заболевания в значительной степени зависят от возраста на момент постановки диагноза и от начала лечения, а также от формы ГФА с дефицитом ВН4 в зависимости от генетически детерминированного дефекта фермента, участвующего в его обмене.
Выделяют шесть форм ГФА, различающихся в отношении подхода к диагностике, лечению и наблюдению:
• ФАГ-дефицитная ГФА, легкая форма (не требующая терапии до года);
2. Диагностика. Несмотря на низкую распространенность дефектов синтеза ВН4, всех новорожденных с ГФА, выявленной при скрининге, необходимо обследовать на дефекты синтеза BH4.
Дефицит BH4 включает группу заболеваний, которые могут выявляться с помощью неонатального скрининга на ГФА, но не м.б. идентифицированы рутинными скрининговыми методами, поэтому селективный скрининг на дефицит BH4 показан каждому новорожденному с уровнем ФА в плазме >120 мкМ/л, а также детям старшего возраста с ГФА и неврологическими симптомами.
Дефицит BH4 и ответственный за нарушение метаболизма дефект фермента м.б. диагностированы с помощью нескольких исследований.
- Измерение неоптерина и биоптерина. В жидкостях организма, преимущественно в моче, измеряют неоптерин (продукт окисления дигидронеоптерина трифосфата) и биоптерин (продукт окисления дигидробиоптерина и BH4) (см. рис. 1). У пациентов с дефицитом ГТФ-циклогидролазы I неоптерин и биоптерин выделяются с мочой в малом количестве. У пациентов с дефицитом 6-пирувоилтетрагидроптеринсинтазы наблюдается значимое повышение экскреции неоптерина и сопутствующее снижение экскреции биоптерина. У пациентов с дефицитом дигидроптеридинредуктазы наблюдается повышенная экскреция неоптерина и биоптерина. Экскреция биоптерина увеличивается при дефиците этого фермента, поскольку хиноноид дигидробиоптерин не может восстановиться обратно в BH4. У пациентов с дефицитом птерин-4α-карбиноламиндегидратазы наблюдается экскреция 7-биоптерина (необычного изомера биоптерина) с мочой.
- Исследования спинномозговой жидкости. Исследование СМЖ позволяет выявить пониженное содержание дофамина и метаболитов серотонина.
- Нагрузочный тест с сапроптерином. Прием сапроптерина (ситнетической формы BH4) внутрь (20 мг/кг) нормализует уровень ФА и соотношение ФА/тирозин в плазме у пациентов с дефицитом BH4 в течение 4-12 ч. Для интерпретации результатов уровень ФА в крови должен быть повышенным (>400 мкмоль/л). Для этого необходимо прекратить диетотерапию за 2 дня до проведения теста/принять нагрузочную дозу ФА (100 мг/кг) за 3 ч до теста. При ВН4-чувствительной ФКУ, вызванной дефицитом ФАГ, содержание ФА в крови может снижаться при проведении нагрузочного теста с ВН4, но в дальнейшем повышаться даже при его добавлении.
Пациенты с уровнем ФА в пределах нормы не <1 нед при отсутствии диеты с ограничением ФА могут продолжить прием ВН4 в качестве единственного средства лечения ГФА. Тем не менее очень важно планово контролировать содержание ФА в плазме для подтверждения его нормального уровня.
Тестирование с сапроптерином проводится >1 года, но возможно повторное проведение теста в более старшем возрасте у больных с ФАГ-зависимой формой ГФА для определения чувствительности к сапроптерину. При этом для оценки нутритивного статуса и функционального состояния внутренних органов рекомендуется контроль развернутого OAK, ОАМ, биохим. анализа сыворотки крови.
- Молекулярное исследование. Секвенирование и анализ делеции/дупликации являются перспективными и доступными методами в практическом 30 и играют все более важную роль в подтверждении биохим. диагноза.
Всем пациентам с клиническим диагнозом ФКУ/ГФА проводится молекулярно-генетическое исследование генов РАН, PTS, QDPR, GCH1, PCBD, SPR, DNAJC12 для верификации клинического диагноза, ДД и выявления степени риска наследственной патологии в семье.
- Ферментный анализ. Активность дигидроптеридинредуктазы можно измерить в пятнах сухой крови на фильтровальной бумаге, используемой для скрининга. Активность 6-пируво-илтетрагидроптеринсинтазы можно измерить в печени, фибробластах и эритроцитах. Активность птерин-4α-карбиноламиндегидратазы можно измерить в печени и фибробластах. Активность ГТФ-циклогидролазы I можно измерить в печени и стимулированных цитокинами (IFN-γ) мононуклеарных клетках/фибробластах (активность фермента в нестимулированных клетках обычно очень низкая).
3. Лечение. Терапия направлена на коррекцию ГФА и устранение дефицита нейромедиаторов в ЦНС. Пациентам с дефицитом кофакторов важно контролировать ГФА, поскольку высокие уровни ФА приводят к задержке умственного развития и препятствуют транспортировке предшественников нейромедиаторов (тирозина и триптофана) в ГМ. Уровень ФА в плазме следует поддерживать как можно ближе к норме (<6 мг/дл/<360 мкмоль/л). Это м.б. достигнуто приемом ВН4 внутрь (5-20 мг/кг в сутки). Сапроптерина гигидрохлорид имеется в продаже, но является дорогостоящим ЛП.
Начальная доза сапроптерина при недостаточности ВН4 составляет 2-5 мг/кг 1 р/сут. Доза м.б. увеличена до 20 мг/кг в сутки, для достижения оптимального терапевтического эффекта суточная доза ЛП м.б. разделена на 2-3 приема.
Большинству этих пациентов необходимо в течение всей жизни принимать БАД предшественников нейромедиаторов, таких как L-ДОФА и 5-гидрркситриптофана, а также карбидопу для ингибирования разложения L-ДОФА до попадания в ЦНС, даже при нормализации уровней ФА в плазме на фоне терапии BH4. BH4 не сразу попадает в ГМ для восстановления синтеза нейромедиаторов. Чтобы свести к минимуму нежелательные побочные явления (особенно ЛП-индуцированную дискинезию, вызванную L-ДОФА), лечение следует начинать с низких доз L-ДОФА/карбидопы и 5-гидрркситриптофана и постепенно корректировать дозу в зависимости от ответа на терапию и клинических результатов в отношении каждого отдельного пациента. Пациентам с дефицитом дигидро-птеридинредуктазы также рекомендуются БАД с фолиевой кислотой.
К сожалению, попытки нормализовать уровни нейромедиаторов с помощью их предшественников обычно не позволяют полностью устранить неврологические симптомы, т.к. не удается достичь нормального уровня BH4 в ГМ. У пациентов часто наблюдаются задержка умственного развития, вариабельные нарушения тонуса, нарушения движений глаз, нарушения равновесия и координации движений, снижение способности к двигательной активности и судороги, несмотря на прием предшественников нейромедиаторов.
У пациентов с дефицитом BH4 возникает гиперпролактинемия, которая м.б. обусловлена дефицитом гипоталамического дофамина. В качестве удобного метода мониторинга достаточного замещения нейромедиаторов у пациентов, имеющих такую патологию, можно проводить измерение уровня пролактина в сыворотке.
Известно, что некоторые ЛП, такие как триметоприм/сульфаметоксазол, метотрексат и др. противолейкемические ЛП, подавляют активность фермента дигидроптеридинредуктазы и поэтому должны применяться с большой осторожностью у пациентов с дефицитом BH4.
Целевые уровни ФА крови — 120-360 мкмоль/л для всех возрастов. Допустимы более высокие колебания ФА крови на диете, но не >600 мкмоль/л. Нижний целевой уровень ФА составляет не <120 мкмоль/л для всех пациентов. У пациентов с BH4-дефицитными формами ГФА целевой уровень ФА <120 мкмоль/л.
4. Генетика и уровень распространения. Все дефекты, вызывающие ГФА, наследуются по АуР-типу. По оценкам, уровень распространения ФКУ в США составляет 1:14 000-20 000 живорождений. Уровень распространения ГФА без ФКУ составляет 1:50 000 живорождений. Заболевание чаще встречается у белых американцев и коренных индейцев, реже у афроамериканцев, латиноамериканцев и азиатов.
В РФ, по данным неонатального скрининга, частота ГФА составляет 1:7000 и варьирует в разл. регионах.
Ген ФАГ расположен в хромосоме 12q23.2, и при исследовании разл. семей было обнаружено множество мутаций, вызывающих это заболевание. У большинства пациентов наблюдаются сложные гетерозиготные состояния по двум разл. мутантным аллелям. Ген 6-пирувоил-тетра-гидроптеринсинтазы (PTS), считающийся наиболее частой причиной дефицита BH4, расположен в хромосоме 1 lq23.1. Ген дигидроптеридинредуктазы (QDPR) расположен в хромосоме 4р15.32, а ген птерин-4α-карбиноламиндегидратазы (PCBD1) и ГТФ-циклогидролазы I (GCH1) расположены в хромосомах 10q22.1 и 14q22.2 соответственно. Пренатальная диагностика возможна в тех случаях, когда причинные мутации известны*.
P.S. * В КР «Классическая фенилкетонурия и другие виды гиперфенилаланинемии» представлена этиопатогенетическая классификация гиперфенилаланинемии.
В качестве профилактики ГФА рекомендуется проспективное медико-генетическое консультирование пар, планирующих беременность, с рекомендацией обследования на гетерозиготное носительство частых мутаций в гене РАН; с целью предупреждения рождения ребенка с синдромом «материнской ФКУ» рекомендуется консультирование девочек подросткового возраста и женщин, страдающих ГФА, по вопросам планирования семьи, поддержание ФА крови на уровне 360 мкмоль/л за 2-3 мес до зачатия и в течение всей беременности, строгие диетические ограничения поступления ФА с пищей, прием BH4 в случае его ранее выявленной эффективности*.
P.S. * КР «Классическая фенилкетонурия и другие виды гиперфенилаланинемии». М., 2020. 112 с. Ассоциация медицинских генетиков; Профессиональная общественная организация (ассоциация) детских врачей «Инициатива специалистов педиатрии и неонатологии в развитии клинических практик»; Союз педиатров России.