При нормальном пути катаболизма метионина, незаменимой аминокислоты, образуется S-аденозилметионин, который служит донором метильной группы для метилирования множества соединений в организме, и цистеин, синтезирующийся в результате ряда реакций, в совокупности носящих название транс-сульфирование (рис. 1).
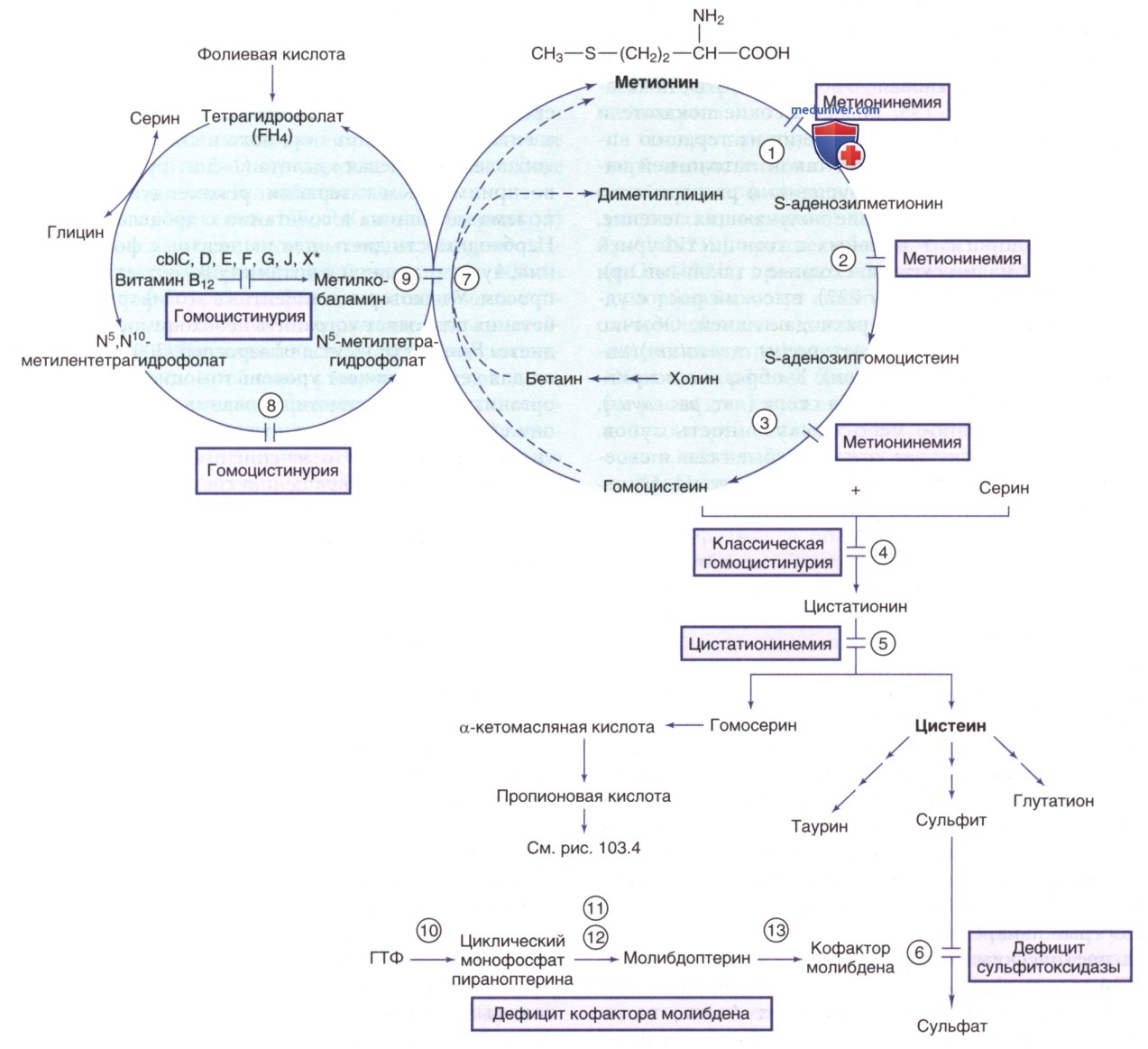
Рисунок 1. Пути метаболизма серосодержащих аминокислот. Ферменты: 1) метионинаденозилтрансфераза (MATI/III); 2) глицин-N-метилтрансфераза, 3) 5-аденозилгомоцистеингидролаза; 4) цистатионинсинтаза; 5) цистатионаза; 6) сульфитоксидаза; 7) бетаингомоцистеинметилтрансфераза; 8) метилентетрагидрофолатредуктаза; 9) метионинсинтаза (cbIG); 10) белок 1 биосинтеза кофактора молибдена; 11) молибдоптеринсинтаза; 12) аденилилтрансфераза и серотрансфераза (MOCS3); 13) гефирин. *Нарушения в кобаламине (cbl) С, D, F, J, X приводят к метилмалоновой ацидемии и гомоцистинурии
а) Гомоцистинурия (гомоцистеинемия). В норме большая часть гомоцистеина, промежуточного соединения разложения метионина, подвергается реметилированию до метионина. Эта метионин-восстанавливающая реакция катализируется ферментом метионинсинтазой, для которого требуется метаболит фолиевой кислоты (5-метилтетрагидрофолат) в качестве донора метила и метаболит витамина В12 (метилкобаламин) в качестве кофактора (см. рис. 1). У ЗЛ большая часть гомоцистеина плазмы связана с белками/существует в виде дисульфидов. Были выявлены три основные формы гомоцистинемии и гомоцистинурии.
1. Гомоцистинурия, вызванная дефицитом цистатионин-β-синтазы (классическая гомоцистинурия). Это наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот. Относится к классу редких (орфанных) заболеваний*.
P.S. * КР РФ «Гомоцистинурия у детей», 2016 г.
Наиболее распространенное врожденное нарушение метаболизма метионина. Примерно 40% пациентов с такой патологией отвечают на терапию высокими дозами витамина В6 и обычно имеют более легкие клинические проявления, чем пациенты, не поддающиеся этому лечению. У таких пациентов наблюдается некоторая остаточная ферментативная активность.
Новорожденные с классической гомоцистинурией при рождении выглядят как обычные здоровые дети. Клинические проявления в младенческом возрасте неспецифичны и могут включать задержку физического и психического развития. Если скрининг новорожденных не проводится, то диагноз ставится позже, обычно >3 лет при подвывихе хрусталика (лат. ectopia lentis). Это приводит к миопии высокой степени и иридодонезу (дрожанию радужной оболочки). В более позднем возрасте могут развиваться астигматизм, глаукома, стафилома, катаракта, отслойка сетчатки и атрофия зрительного нерва. Часто возникает прогрессирующая умственная отсталость. Также зарегистрированы случаи с сохранением нормального интеллекта.
По данным международного опроса, в котором участвовало >600 пациентов, показатели IQ составляли 10-135. Более высокие показатели наблюдаются у пациентов, отвечающих на терапию витамином В6. У >50% пациентов с такой патологией наблюдались психиатрические нарушения и расстройство поведения. У 20% пациентов, не получающих лечение, наблюдаются судороги. У больных с гомоцистинурией наблюдаются аномалии скелета, сходные с таковыми при синдроме Марфана (Marfan): высокий рост с удлиненными конечностями и арахнодактилией.
Обычно наблюдаются сколиоз, впалая (лат. pectus excavatum)/килевидная ГК (лат. pectus carinatum), Х-образное искривление ног (лат. genu valgum), полая стопа (лат. pes cavus), высокое куполообразное нёбо и скученность зубов. У этих детей обычно светлая кожа, голубые глаза и своеобразный румянец на скулах. Основным рентгенологическим признаком является генерализованный остеопороз, особенно позвоночника.
У детей старшего возраста часто бывают переломы. Наряду с этим встречаются формы болезни, при которых изменения опорно-двигательного аппарата минималь-ны/полностью отсутствуют.
ТЭО с вовлечением крупных/мелких сосудов, в особенности сосудов ГМ, являются обычным явлением и могут возникать в любом возрасте. ТЭО, вероятно, вызвана повышенным уровнем гомоцистеина, приводящим к аномальному ангиогенезу и ингибированию фибринолитической активности. К числу ее серьезных последствий относятся атрофия зрительного нерва, паралич, легочное сердце и тяжелая АГ (вследствие инфаркта почек). Риск ТЭО увеличивается после хирургических вмешательств. К редким осложнениям относятся спонтанный пневмоторакс и острый панкреатит.
Диагностическим лабораторным признаком является повышение содержания метионина и гомоцистина (или гомоцистеина) в физиол. жидкостях. Анализ на гомоцистин необходимо проводить в свежей порции мочи, поскольку это соединение нестабильно и при длительном хранении может разрушиться. В плазме наблюдается низкий уровень цистеина/полное его отсутствие. Концентрация гомоцистеина и гомоцистина в плазме определяется после двух недель отмены пиридоксина (в т.ч. в составе поливитаминов)4.
Для контроля лечения предпочтение отдается анализу на общий гомоцистеин плазмы. Свободный гомоцистеин в плазме может нормализоваться/оставаться нормальным при снижении общего гомоцистеина плазмы. Диагноз м.б. установлен молекулярным анализом на цистатионин-β-синтазу/анализом фермента в культуре фибробластов, лимфоцитов, стимулированных фитогемагглютинином, и биоптатах печени.
После подтверждения диагноза гомоцистинурии вследствие недостаточности фермента цистатион-β-синтазы возможно проведение теста с пиридоксином; его задача — ДД двух фенотипических вариантов классической гомоцистинурии: В6-зависимого и В6-резистентного.
Лечение высокими дозами витамина В6 (100-500 мг/ сут) вызывает значительное улучшение у пациентов, отвечающих на терапию. Степень ответа на лечение может варьировать в зависимости от семьи. Некоторые пациенты могут не отвечать на терапию из-за низкого уровня фолиевой кислоты; нельзя считать пациента невосприимчивым к витамину В6 до тех пор, пока к схеме лечения не будет добавлена фолиевая кислота (1-5 мг/сут). Пациентам, невосприимчивым к терапии, рекомендуется ограничение приема метионина в сочетании с добавлением цистеина. Необходимость диеты для пациентов с формой заболевания, чувствительной к витамину В6, остается спорным вопросом.
У некоторых пациентов с этой формой добавление бетаина позволяет устранить необходимость в какой-либо диете. Бетаин (6 г/сут для взрослых/200-250 мг/кг в сутки для детей) снижает уровень гомоцистеина в жидкостях организма за счет реметилирования гомоцистеина в метионин (см. рис. 1), что позволяет повысить уровень метионина в плазме. Это лечение приводит к клиническому улучшению (предотвращению сосудистых осложнений) у пациентов, не отвечающих на терапию витамином В6. У пациента с нечувствительной к витамину В6 гомоцистинурией, не соблюдавшего диету при терапии бетаином, может развиться ОГМ.
Диетическое лечение заключается в снижении потребления метионина за счет ограничения потребления белка натуральных продуктов. Для предотвращения белково-энергетической недостаточности назначаются специализированные продукты питания.
Зарегистрировано >100 случаев беременности у женщин с классической гомоцистинурией с благоприятными исходами как для матерей, так и для новорожденных. Большинство детей рождались доношенными и здоровыми. У нескольких матерей возникли послеродовые ТЭО.
Скрининг новорожденных на классическую гомоцистинурию проводится во всем мире. По оценкам, заболевание встречается в 1:200 000-350 000 живорожденных, хотя в некоторых частях мира оно м.б. более распространенным (напр., 1:1800 в Катаре). Раннее лечение пациентов, выявленных в результате скрининга, способствует благоприятному исходу. У пациентов с нечувствительной к витамину В6 формой заболевания, получавших лечение в раннем возрасте, средний IQ находился в пределах нормы. У некоторых пациентов лечение предотвращало развитие эктопии хрусталика.
Классическая гомоцистинурия наследуется как АуР-признак. Ген цистатионин-β-синтазы (CBS) расположен в хромосоме 21q22.3. Пренатальную диагностику проводят методом ДНК-анализа/ферментативного анализа культуры амниотических клеток. Большинство пациентов с таким нарушением являются сложными гетерозиготами по двум разл. аллелям. У гетерозиготных носителей симптомы отсутствуют.
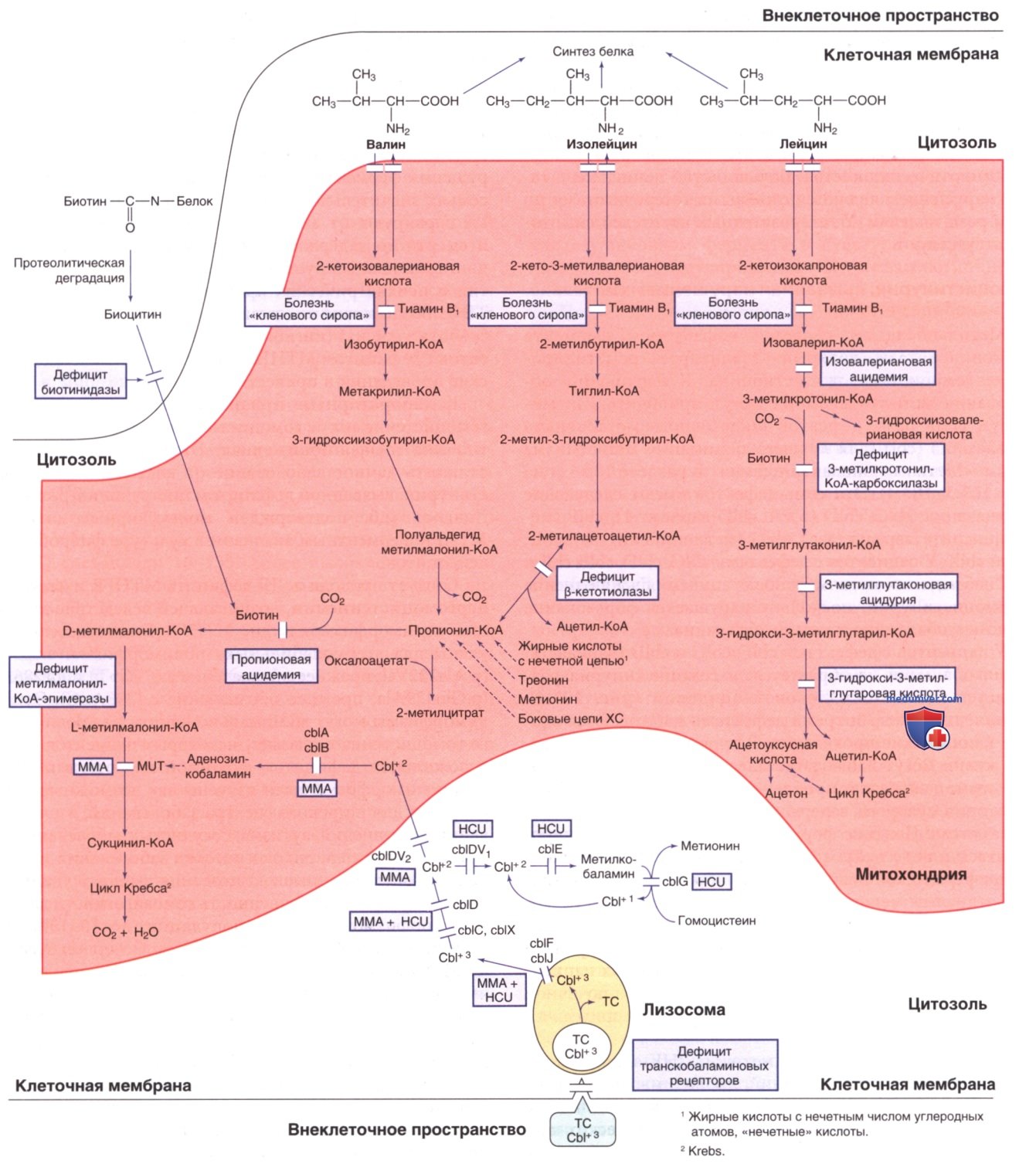
2. Гомоцистинурия, вызванная нарушением синтеза метилкобаламина. Метилкобаламин является кофактором фермента метионинсинтазы, который катализирует реметилирование гомоцистеина до метионина. К нарушению образования метилкобаламина могут приводить как минимум семь разл. дефектов в/клеточного метаболизма кобаламина (cbl) (для лучшего понимания метаболизма cbl см. «Метилмалоновая ацидемия» в отдельной статье на сайте - просим пользоваться поиском по сайту выше). Эти семь дефектов имеют следующие обозначения: сЫС, cblD (в т.ч. cblD вариант 1), сЫЕ (метионинсинтетазредуктаза), cblG (метионинсинтаза), cblF, cblJ и cblХ.
У пациентов с дефектами cb1С, cblD, cblF, cblJ и cblХ наблюдается метилмалоновая ацидемия в дополнение к гомоцистинурии, поскольку нарушается образование аденозилкобаламина и метилкобаламина.
У пациентов с дефектами cblЕ, cblG и cblD (вариант 1) метилкобаламин не образуется, и гомоцистинурия развивается без метилмалоновой ацидемии (рис. 2). У пациентов с этими тремя дефектами наблюдаются схожие клинические проявления. В первые несколько месяцев жизни могут наблюдаться неспецифические симптомы, такие как рвота, проблемы с кормлением, вялость, гипотония, судороги, задержка физического и психического развития. Поздние формы этих патологий могут проявляться в виде нейрокогнитивных нарушений, психоза и периферической невропатии. К лабораторным признакам относятся мегалобластная анемия, гипергомоцистеи-немия, гомоцистинурия и гипометионинемия.
Рисунок 2. Пути метаболизма аминокислот с разветвленной цепью, биотина и витамина В12 (кобаламина). Cbl — кобаламин; cbl — нарушение метаболизма кобаламина; cblDV1 — кобаламин D (вариант 1); cblDV2 — кобаламин D (вариант 2); HCU — гомоцистинурия; ММА — метилмалоновая ацидемия; MUT — мутаза; OHcbl — гидроксокобаламин; ТС — транскобаламин;TCR — рецептор транскобаламина. Названия ферментов приведены в тексте
Отсутствие гиперметионинемии отличает эти состояния от дефицита цистатионин-|3-синтазы (см. ранее). У некоторых пациентов с этими нарушениями отмечались тромбоз почечной артерии, ГУС, легочная гипертензия и атрофия зрительного нерва.
Диагноз устанавливается методом ДНК-анализа/комплементационных исследований, проводимых в культуре фибробластов. Пренатальную диагностику выполняют с помощью исследований на культурах амниотических клеток. Дефициты cblЕ, cblG и cblD (вариант 1) наследуются как АуР-признаки. Геном сЫЕ является MTRR, кодирующий метионинсинтетазредуктазу (расположен в хромосоме 5р15.31). Геном cblG является MTR, кодирующий метионинсинтазу (расположен в хромосоме lq43). Дефицит cblD варианта 1 вызван патогенными вариантами, влияющими на С-концевой ген MMADHC (расположен в хромосоме 2q23.2).
Лечение витамином В12 в виде высоких доз гидроксокобаламина помогает улучшить клинические и биохим. показатели. Результаты варьируют в зависимости от формы заболевания и от родства.
3. Гомоцистинурия, вызванная дефицитом метилентетрагидрофолатредуктазы (дефицит MTHFR). Этот фермент восстанавливает 5,10-метилентетрагидрофолат с образованием 5-метилтетрагидрофолата, который отдает метильную группу для реметилирования гомоцистеина до метионина (см. рис. 1). Степень нарушения фермента и клинические проявления в разных семьях значительно различаются. Клинические признаки варьируют от апноэ, судорог, микроцефалии, комы и смерти до задержки психического развития, атаксии, двигательных нарушений, периферической невропатии и психиатрических проявлений. Также наблюдалась ТЭО. Воздействие анестезирующего динитрогена оксида («Азота закиси») (ингибирует метионинсинтазу) на пациентов с дефицитом MTHFR может ухудшить неврологические проявления и привести к смерти.
К лабораторным признакам относятся умеренная гомоцистеинемия и гомоцистинурия. Концентрация метионина низкая/пониженная. Этот признак позволяет отличить данное заболевание от классической гомоцистинурии, вызванной дефицитом цистатионин-р-синтазы. Диагноз м.б. подтвержден молекулярным анализом MTHFR/ферментным анализом в культуре фибробластов/ лейкоцитов.
Следует проводить ДД дефицита MTHFR и легкой гипергомоцистеинемии, возникающей вследствие двух общих полиморфизмов в гене MTHFR. Были тщательно изучены два «термолабильных» полиморфизма: с.665С>Т (p.Ala222Val, прежнее обозначение с.677С>Т) и с.1286А>С (p.Glu429Ala, прежнее обозначение с.1298А>С). Эти полиморфизмы могут минимально влиять на уровни общего гомоцистеина в плазме у некоторых пациентов и часто осложняются дефицитом фолиевой кислоты в питании.
Оба полиморфизма были изучены как возможные факторы риска для широкого спектра заболеваний, в т.ч. врожденных нарушений, аутизма, сосудистых заболеваний, инсульта, выкидыша, онкологических заболеваний и ответа на XT. Популяционные исследования выявили удивительно высокую распространенность гомозиготности для этих полиморфизмов в общей популяции: до 10-15% представителей белой европеоидной расы Северной Америки и >25% у некоторых латиноамериканцев.
Предполагается, что обогащение муки фолиевой кислотой способствует снижению степени взаимосвязей, наблюдавшихся ранее. Современные данные подтверждают роль полиморфизма с.665С>Т (ранее с.677С>Т) как фактора риска дефектов нервной трубки. Несмотря на доступность клинических тестов на этот полиморфизм, недавние метаанализы не подтвердили связь между полиморфизмом MTHFR и риском венозной тромбоэмболии, а также между легкой формой гипергомоцистеинемии и повышенным риском ИБС.
Заболевание наследуется по АуР-типу. Диагноз м.б. подтвержден анализом гена MTHFR. Пренатальная диагностика может выполняться методом молекулярного анализа MTHFR известных семейных патогенных вари-антов/путем измерения активности фермента MTHFR в культурах клеток хориальных ворсинок/амниоцитов.
Предпринималась попытка лечения дефицита MTHFR комбинацией фолиевой кислоты, витамина В6, витамина В12, БАД метионина и бетаина. Раннее лечение бетаином дает наиболее «+» эффект.
б) Гиперметионинемия:
1. Первичная (генетическая) гиперметионинемия. Повышение уровня метионина в плазме наблюдается при нескольких генетических заболеваниях.
- Классическая гомоцистинурия. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
- Дефицит метионинаденозилтрансферазы печени (болезнь Мадда (Mudd). Этот фермент имеет две изоформы (тетрамерную MAT I и димерную MAT III), кодируется одним геном (МАГМ в хромосоме 10q22.3) и участвует в 1-й стадии катаболизма метионина (см. рис. 1). Др. фермент (MAT II), сходный по структуре, кодируется др. геном (МАТ2А в хромосоме 2р11.2) и экспрессируется преимущественно в непеченочных тканях (почки, ГМ, лимфоциты). Дефицит MAT I/MAT III вызывает гиперметионинемию. При тяжелом дефиците также м.б. повышен общий гомоцистеин в плазме. Большинство пациентов с такой патологией были выявлены в неонатальном периоде в ходе скрининга на гомоцистинурию.
У большинства пациентов наблюдается остаточная ферментативная активность, и они остаются бессимптомными в течение всей жизни, несмотря на стойкую гиперметионинемию. Некоторые жалуются на необычный запах изо рта, вероятно, вызванный накоплением диметилсульфида. У нескольких пациентов с полным дефицитом ферментов отмечались неврологические отклонения, обусловленные демиелинизацией (умственная отсталость, дистония, диспраксия).
При лабораторных исследованиях наблюдаются заметно повышенные уровни метионина в плазме при нормальном/низком уровне S-аденозилметионина и нормальных концентрациях S-аденозилгомоцистеина и гомоцистеина. Указанные признаки позволяют провести ДД дефицит MAT I/MAT III от др. причин гиперметионинемии.
На данный момент нет общепринятой схемы лечения. Если пациентам не назначается специфическое лечение, необходима организация длительного наблюдения для выявления неврологических нарушений и аномалий функции печени. Диеты с низким содержанием метионина понижают уровень метионина в плазме, однако целесообразность таких диет является спорным вопросом, поскольку снижение уровня метионина в плазме вызывает дальнейшее снижение уровня S-аденозилметионина в организме. Прием БАД S-аденозилметионина в сочетании с диетой с низким содержанием метионина кажется обоснованным, но пока отсутствует большой клинический опыт.
Среди матерей с дефицитом MAT I/MAT III (МАТ1А) зарегистрированы нормальные беременности с рождением здорового ребенка. Заболевание наследуется по АуР-типу, хотя патогенный вариант p.R264H в МАТ1А, по всей видимости, нарушает димеризацию белка и может привести к легкой гиперметионинемии даже у гетерозиготных пациентов.
- Дефицит глицин-N-метилтрансферазы. Глицин-N-метилтрансфераза способствует катаболизму S-аденозилметионина в S-аденозилгомоцистеин (см. рис. 1). В настоящее время зарегистрировано несколько пациентов с дефицитом этого фермента. Пациенты были клинически бессимптомными, за исключением легкой гепатомегалии и повышенных уровней трансаминаз в сыворотке крови. Др. лабораторные данные включали гиперметионинемию и очень высокий уровень S-аденозилметионина в сыворотке. Специфическое лечение пока не разработано. Заболевание наследуется как АуР-признак; ген GNMT находится в хромосоме 6р21.1.
- Дефицит S-аденозилгомоцистеингидролазы. Описание дефицита S-аденозилгомоцистеингидролазы (SAHH) (см. рис. 1) встречается нечасто. Общими клиническими проявлениями были умственная отсталость, тяжелая гипотония и прогрессирующая дисфункция печени. При лабораторных исследованиях наблюдался повышенный уровень КФК в сыворотке крови, гипоальбуминемия (вызвавшая водянку плода в одной семье), гипопротромбинемия и значительно повышенный уровень S-аденозилгомоцистеина в сыворотке с умеренным повышением метионина и S-аденозилметионина в плазме. Считается, что заметное повышение уровня S-аденозилгомоцистеина приводит к ингибированию метилтрансфераз, в т.ч. участвующих в синтезе креатина (см. рис. 3) и холина, что приводит к их дефициту. Замедленную миелинизацию белого в-ва можно выявить при проведении МРТ ГМ.
Диагноз м.б. поставлен путем анализа гена AHCY (хромосома 20q11.22)/биохим. анализа эритроцитов, культуры фибробластов кожи и биоптатов печени. В качестве лечения использовалась диета с низким содержанием метионина, но ее долгосрочная эффективность не установлена.
- Тирозинемия типа I. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
- Дефицит цитрина. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
2. Приобретенная (негенетическая) гиперметионинемия. Гиперметионинемия возникает у недоношенных и некоторых доношенных детей, получающих диету с высоким содержанием белка, и, вероятно, отражает замедленное созревание фермента МАТ. Аномалия обычно устраняется при снижении потребления белка. Это нарушение часто встречается у пациентов с разл. формами заболеваний печени.
в) Первичная цистатионинемия (цистатионинурия). Дефицит цистатионазы (цистатионин-γ-лиазы) приводит к значительной цистатионинурии и легкой/умеренной цистатионинемии. Дефицит этого фермента наследуется по АуР-типу и, согласно оценкам, встречается в 1:14 000 живорожденных. Зарегистрирован широкий спектр клинических проявлений. Отсутствие последовательной клинической картины и наличие цистатионинурии у ряда лиц, не имеющих клинических признаков, позволяют предположить, что дефицит цистатионазы может не иметь клинического значения. Во многих случаях зарегистрирован ответ на прием внутрь больших доз витамина В6 (>100 мг/сут).
При выявлении цистатионинурии пациенту можно назначить лечение витамином В6, но его «+» эффект не установлен. Ген, кодирующий цистатионазу (СТН), расположен в хромосоме 1р31.1. Заболевание наследуется по АуР-типу.
Первичную цистатионинурию необходимо отличать от вторичной, которая может возникать у пациентов с дефицитом витаминов В6/В12, заболеванием печени (особенно повреждением, вызванным галактоземией), тиреотоксикозом, гепатобластомой, нейробластомой, ганглиобластомой и дефектами реметилирования гомоцистеина.