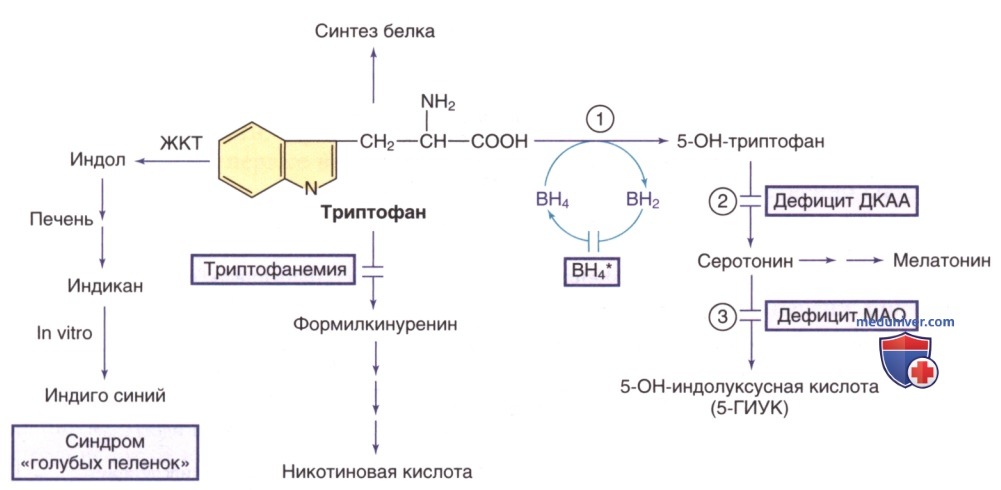
Триптофан является незаменимой аминокислотой и предшественником никотиновой кислоты (ниацина) и серотонина (рис. ниже). Генетические нарушения метаболизма серотонина (одного из основных нейромедиаторов) обсуждаются в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше.
Пути метаболизма триптофана. Тетрагидробиоптерин (ВН4*) указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. ниже). Ферменты: 1) триптофангидроксилаза; 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) МАО
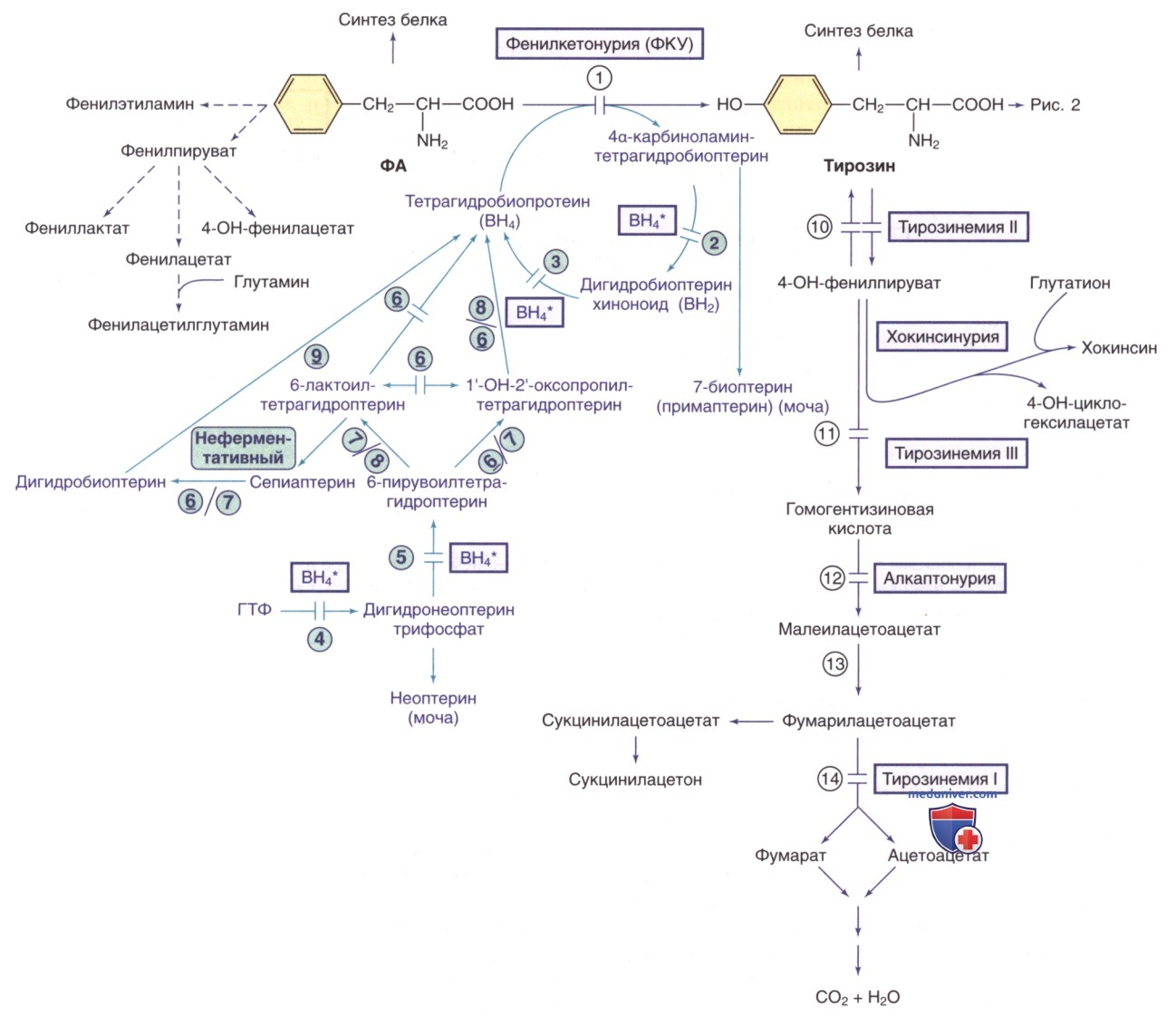
Пути метаболизма фенилаланина (ФА) и тирозина. Генетически детерминированные ферментативные нарушения показаны горизонтальными линиями, пересекающими стрелку(-и) реакции. Пути синтеза кофактора тетрагидробиоптерина (ВН4) показаны фиолетовым цветом. ВН4* относится к дефектам метаболизма ВН4, влияющим на гидроксилазы ФА, тирозина и триптофана (рис. ниже и выше). Ферменты: 1) фенилаланингидроксилаза; 2) птеринкарбиноламиндегидратаза; 3) дигидробиоптеринредуктаза; 4) гуанозинтрифосфат (ГТФ)-циклогидролаза; 5) 6-пирувоилтетрагидроптеринсинтаза; 6) сепиаптеринредуктаза; 7) карбонилредуктаза; 8) альдолазоредуктаза; 9) дигидрофолатредуктаза; 10) тирозинаминотрансфераза; 11) 4-гидроксифенилпируватдиоксигеназа; 12) диоксигеназа гомогентизиновой кислоты; 13) малеилацетоацетат изомераза; 14) фумарилацетоацетат гидролаза
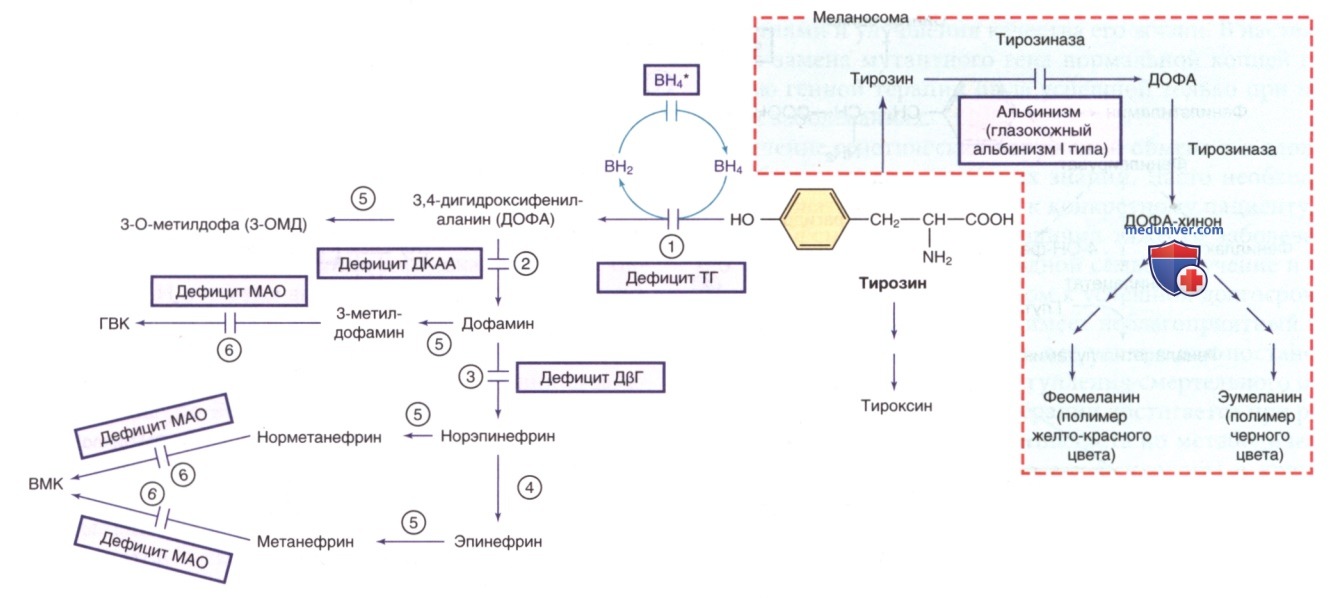
Другие пути метаболизма тирозина. ВН4* указывает на гиперфенилаланинемию, вызванную дефицитом ВН4 (см. рис. 1). ГВК — го-мованиловая кислота; ВМК — ванилилминдальная кислота; ВН4 — тетрагидробиоптерин. Ферменты: 1) тирозингидроксилаза (ТГ); 2) декарбоксилаза ароматических L-аминокислот (ДКАА); 3) дофамин-β-гидроксилаза (ДβГ); 4) фенилэтаноламин-N-метилтрансфераза (ФНМТ); 5) катехол-О-метилтрансфераза (КОМТ); 6) МАО
а) Болезнь Хартнупа. При АуР-болезни Хартнупа (Hartnup), названной в честь первой заболевшей семьи, в слизистой оболочке кишечника и почечных канальцах возникает нарушение транспорта моноаминомонокарбоновых аминокислот (нейтральных аминокислот), в т.ч. триптофана. Транспортный белок этих аминокислот (В°АТ1) кодируется геном SLC6A19 (расположен в хромосоме 5р15.33). В большинстве случаев клинические симптомы заболевания отсутствуют.
Проявление болезни у пациентов сильно варьирует. Вероятно, это связано с особенностями питания, окружающей среды и генетической гетерогенностью (напр., два белка, ТМЕМ27 и АСЕ2, взаимодействующие с В°АТ1). Снижение всасывания триптофана в кишечнике в сочетании с его повышенными ренальными потерями может привести к уменьшению количества триптофана, участвующего в синтезе ниацина. Дефицит триптофана может усугубляться вследствие синдрома мальабсорбции, напр. при целиакии. Основным клиническим проявлением у пациентов с редкими симптомами является светочувствительность кожи.
Даже после недолгого пребывания на солнце кожа грубеет и краснеет, а при более длительном воздействии может развиться сыпь наподобие пеллагры. Сыпь может сопровождаться зудом, может развиться хроническая экзема. У новорожденных с этой патологией в возрасте 10 дней наблюдались изменения кожи. У некоторых пациентов может развиться перемежающаяся атаксия, проявляющаяся неустойчивой походкой с широко расставленными ногами. Атаксия может длиться несколько дней, и состояние улучшается при приеме никотиновой кислоты («Ниацина»). Когнитивные функции обычно в пределах нормы.
Наблюдались эпизодические психиатрические нарушения, такие как повышенная возбудимость, эмоциональная нестабильность, депрессия и суицидальные наклонности; эти изменения обычно сопутствуют приступам атаксии. У некоторых пациентов наблюдались низкорослость и атрофический глоссит.
У большинства детей с диагностированной в результате неонатального скрининга болезнью Хартнупа симптомы отсутствуют. Это указывает на вовлечение в патогенез и др. факторов.
Основным лабораторным признаком является аминоацидурия, которая представлена нейтральными аминокислотами (аланин, серин, треонин, валин, лейцин, изолейцин, ФА, тирозин, триптофан, гистидин). Экскреция с мочой пролина, гидроксипролина и аргинина остается в норме. По этому признаку болезнь Хартнупа можно отличить от др. причин генерализованной аминоацидурии, напр. от синдрома Фанкони. Концентрация нейтральных аминокислот в плазме нормальная/незначительно снижена. Этот неожиданный на первый взгляд признак отражает компенсаторные механизмы, требуемые для поддержания нормального транспорта и использования аминокислот.
У некоторых пациентов м.б. обнаружено большое количество производных индола (особенно индикана), являющееся следствием бактериального разложения неабсорбированного триптофана в кишечнике.
Болезнь Хартнупа диагностируется на основании четкой периодичности возникающих симптомов и характерных результатов анализа аминокислот в моче. При необходимости диагноз м.б. подтвержден молекулярным анализом гена SLC6A19.
Пациенты с симптомами болезни Хартнупа отвечают на лечение никотиновой кислотой/никотинамидом (50-300 мг/сут), а также на диету с высоким содержанием белка. Из-за непостоянства клинических проявлений эффективность такого лечения трудно оценить. Распространенность синдрома Хартнупа составляет 1:20 000-55 000 живорожденных. У нескольких женщин с этим заболеванием зарегистрирован благоприятный исход беременности — как для матери, так и для плода.