Синдром Марфана (Marfan syndrome) — это наследственное СЗСТ, вызванное мутациями в гене, кодирующем белок внеклеточного матрикса фибриллин-1. В первую очередь заболевание проявляется патологией скелета, ССС и глаз. Диагноз ставится на основании клинических данных, некоторые из которых зависят от возраста.
а) Эпидемиология. Заболеваемость составляет 1:10 000 живорождений, и 25% случаев являются спорадическими. Расстройство характеризуется АуД-наследованием с высокой пенетрантностью, но вариабельной экспрессией; распространены как межсемейные, так и в/семейные клинические вариации. Расовых и гендерных особенностей не отмечается.
б) Патогенез. Синдром Марфана обусловлен аномальной продукцией, отложением в матриксе и/или стабильностью фибриллина-1 — белка внеклеточного матрикса массой 350 кДа, который является основным компонентом микрофибрилл, с выраженным разрушением микрофибрилл и эластических волокон в пораженных тканях. Локус фибриллина-1 (FBN1) расположен на длинном плече хромосомы 15 (15q21), ген состоит из 65 экзонов.
Анализ сцепления показал отсутствие гетерогенности локуса, FBN1 участвует в >90% случаев, на сегодняшний день выявлено >1000 вызывающих заболевание мутаций (большинство из которых представляют собой точечные миссенс-мутации, уникальные для данной семьи). За исключением ранних и тяжелых форм заболевания, вызванных некоторыми мутациями в экзонах 26-27 и 31-32, четкой корреляции между генотипом и фенотипом не выявлено. Учитывая значительную в/семейную вариабельность, генетические, эпигенетические, экологические и др. неустановленные факторы могут влиять на проявление заболевания.
Традиционно считалось, что синдром Марфана является результатом структурного дефицита соединительной ткани. Считалось, что снижение уровня фибриллина-1 приводит к первичному нарушению отложения эластичных волокон, т.к. и в коже, и в аорте пациентов отмечается снижение количества эластина наряду с фрагментацией эластических волокон. Считалось, что в ответ на стресс (напр., воздействие гемодинамических сил в проксимальном отделе аорты) в пораженных органах реакцией на эту структурную недостаточность будет ускоренная дегенерация.
Однако было трудно согласовать некоторые проявления заболевания, напр., разрастание костей, черепно-лицевой дисморфизм и снижение мышечной массы/ жировых запасов, с этой моделью структурного дефицита.
Семейство цитокинов трансформирующего фактора роста β (TGF-β) влияет на разнообразные клеточные процессы, включая пролиферацию, миграцию, дифференцировку, выживание и синтетическую активность. TGF-β-лиганды (TGF-β1, TGF-β2, TGF-β3) синтезируются в виде неактивных комплексов-предшественников и секвестрируются белками внеклеточного матрикса, в т.ч. фибриллином-1.
У мышей, гетерозиготных по мутации в гене фибриллина-1, в типичном случае вызывающей синдром Марфана у людей, отмечаются многие классические признаки этого заболевания, включая аневризму корня аорты с тканевой характеристикой повышенной передачи сигналов TGF-β, это позволяет предположить, что неудачная секвестрация внеклеточным матриксом с помощью фибриллина-1 латентного TGF-β приводит к увеличению активации и передачи сигнала TGF-β. Кроме того, в мышиных моделях синдрома фармакологическое подавление передачи сигналов TGF-β уменьшает аневризму аорты, доказывая, что повышенная передача сигналов TGF-β является причиной, а не следствием прогрессирования заболевания.
Аберрантная передача сигналов TGF-β также может играть роль в более широком спектре проявлений синдрома Марфана. У мышей с синдромом Марфана наблюдалась повышенная передача сигналов TGF-β и в др. тканях, включая развивающееся легкое, митральный клапан и скелетные мышцы. Лечение этих мышей антагонистами TGF-β уменьшает/предотвращает эмфизему легких, миксоматозную дегенерацию митрального клапана и миопатию скелетных мышц.
Важная роль нарушения регуляции TGF-β в патогенезе синдрома Марфана дополнительно подтверждается открытием и описанием др. родственного синдрома с аневризмой аорты — синдрома Лойса'-Дитца1 2, при котором у пациентов имеют место мутации рецепторов TGF-β и много общих клинических черт с синдромом Марфана (см. раздел «Дифференциальный диагноз»). Дополнительным подтверждением данного факта является синдром Шпринтцена3-Голдберга4, демонстрирующий фенотипическую схожесть как с синдромом Марфана, так и с синдромом Лойса-Дитца, который вызван мутациями в SKI — известном репрессоре сигнального пути TGF-β.
в) Клинические проявления. Синдром Марфана — мультисистемное заболевание с основной симптоматикой со стороны скелета, ССС и глаз.
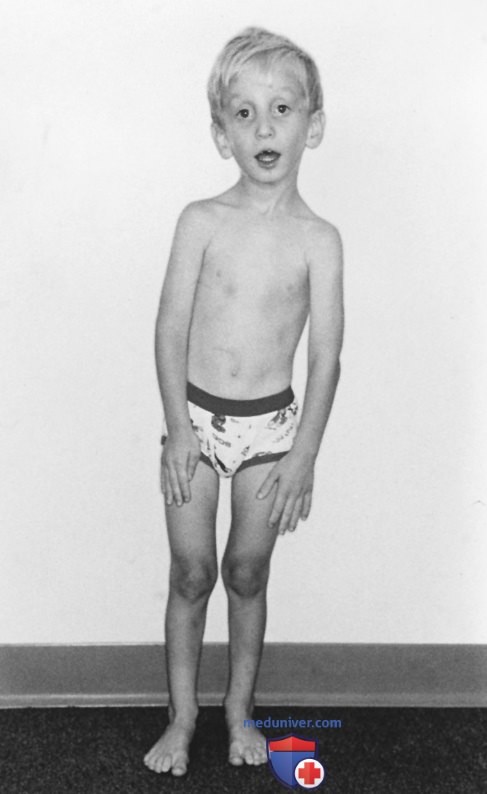
1. Скелет. Чрезмерный рост длинных костей (долихостеномелия) часто является наиболее очевидным проявлением синдрома Марфана и может приводить к уменьшению соотношения верхнего и нижнего сегментов/отношения размаха рук к росту >1,05 раза. Аномальные соотношения верхнего и нижнего сегментов: <1 для возраста 0-5 лет, <0,95 для возраста 6-7 лет, <0,9 для возраста 8-9 лет и <0,85 для возраста >10 лет. Деформация переднего отдела ГК, вероятно, является результатом чрезмерного роста ребер с выпячиванием грудины кнаружи (pectus carinatum, килевидная деформация ГК)/внутрь (pectus excavatum, воронкообразная деформация ГК).
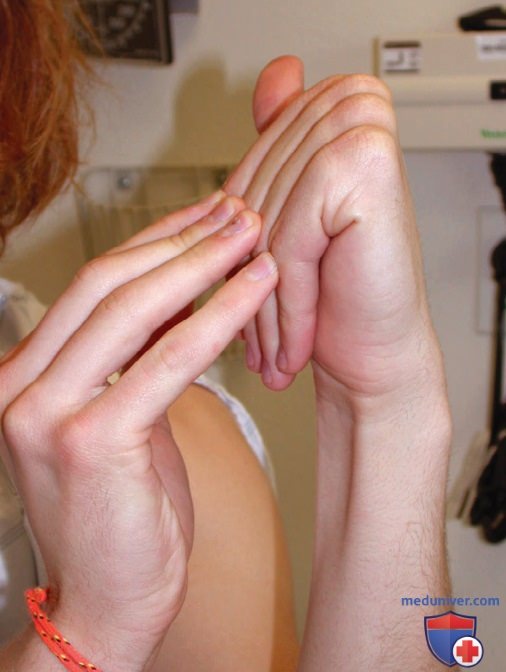
Результатом ускоренного роста позвонков частично м.б. аномальные искривления позвоночника (чаще всего грудопоясничный сколиоз). Др. особенности скелета включают протрузию вертлужной впадины в полость таза (protrusio acetabuli), плоскостопие (pes planus) и гипермобильность (рис. 1) и контрактуры суставов. Длинные и тонкие пальцы по отношению к размеру ладони (арахнодактилия) — субъективный признак.
Рисунок 1. Слабость суставов у пациента с синдромом Марфана
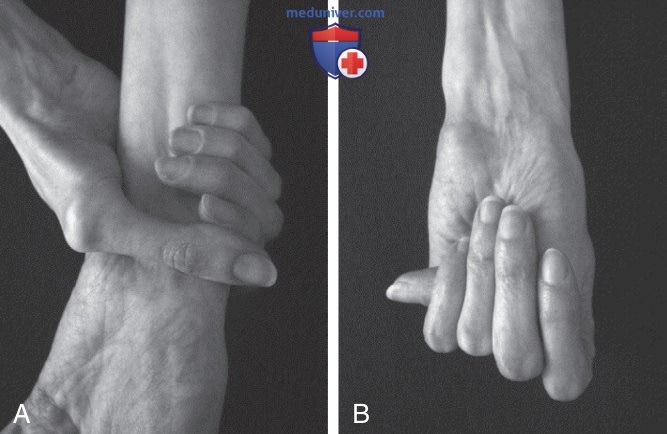
Сочетание арахнодактилии и гипермобильности суставов определяется по наличию знака Уокера-Мердока (Walker, Murdoch)/знака запястья, который считается положительным при полном перекрытии дистальных фаланг большого и пятого пальцев при обхвате контралатерального запястья (рис. 2), а также знака Штейнберга (Sign Steinberg)/знака большого пальца, который имеет место, если дистальная фаланга большого пальца полностью выходит за локтевой край кисти при сгибании его поперек ладони (см. рис. 2).
Обычно наблюдается контрактура пальцев рук (камптодактилия) и локтевых суставов. Могут отмечаться разл. черепно-лицевые аномалии, включая длинный узкий череп (долихоцефалия), глубоко посаженные глаза (энофтальм), смещение кзади нижней челюсти (ретрогнатия)/маленький подбородок (микрогнатия), уплощение средней части лица (скуловая гипоплазия), высокое сводчатое нёбо и наклон глазных щелей книзу (рис. 3).
Рисунок 2. A — знак запястья (или запястье Уокера-Мердока). При обхвате запястья противоположной рукой большой палец перекрывает концевую фалангу пятого пальца; B — знак большого пальца (или большой палец Штейнберга). При сжатии кисти в кулак без посторонней помощи весь ноготь большого пальца выступает за пределы ладони
Рисунок 3. Синдром Марфана. Обратите внимание на удлиненное лицо, опущенные веки, выраженную долихостеномелию и сколиоз легкой степени
2. Сердечно-сосудистая система. Утолщение АВ-клапанов — явление частое, обычно сопровождается их пролапсом. Может иметь место разл. степень регургитации. У детей с ранним началом и тяжелой формой синдрома Марфана недостаточность митрального клапана может привести к СН, легочной АГ и смерти в младенчестве; эта форма является основной причиной заболеваемости и смертности детей раннего возраста с этим заболеванием. Суправентрикулярные аритмии могут наблюдаться в сочетании с дисфункцией митрального клапана. У детей с синдромом Марфана также описываются желудочковые аритмии, наблюдается повышенная распространенность удлиненного интервала Q-T.
У пациентов с этим заболеванием встречается ДКМП, что чаще всего обусловлено перегрузкой объемом, вызванной клапанной регургитацией. Дисфункция аортального клапана, как правило, возникает поздно и обусловлена растяжением аортального кольца расширяющейся аневризмой корня аорты.
Аневризма, расслоение и разрыв аорты, главным образом на уровне синусов Вальсальвы (также известных как корень аорты), остаются наиболее опасными для жизни проявлениями синдрома Марфана, требующими пожизненного мониторинга с помощью ЭхоКГ и др. методов визуализации. В тяжелых случаях аневризмы могут формироваться в/утробно, но в легких они могут отсутствовать/никогда не превышать размеры, требующие клинического вмешательства. Размеры аорты следует интерпретировать в соответствии с возрастными номограммами.
Наиболее важными факторами риска расслоения аорты являются максимальный размер корня аорты и положительный семейный анамнез. Характерными гист. признаками со стороны аорты у пациентов с синдромом Марфана являются кистозный медиальный некроз средней оболочки и разрушение эластичных пластинок. Кистозный медиальный некроз характеризуется очаговым апоптозом, исчезновением сосудистых гладкомышечных клеток и эластических волокон в средней оболочке стенки аорты с последующим отложением муциноподобного в-ва в кистозном пространстве. Эти изменения приводят к утолщению, снижению растяжимости и увеличению жесткости аорты, которая более склонна к расслоению.
У большинства пациентов с острым расслоением аорты наблюдаются классические симптомы, в т.ч. внезапно возникающая сильная, разрывающая боль в груди, часто иррадиирующая в спину.
Расслоение обычно начинается в корне аорты и может ограничиваться восходящим отделом (тип II)/спускаться в нисходящий отдел (тип I). При нарушении функции аортального клапана может развиться острая СН, а также нарушение мозгового кровообращения при вовлечении сонных артерий. Вовлечение КА может предвещать внезапную сердечную смерть, вторичную относительно ИМ/разрыва перикардиальной сумки с последующей тампонадой перикарда. Хроническое расслоение аорты обычно протекает более незаметно, часто без боли в груди. Расширение легочного ствола — частое явление, но обычно не вызывает каких-либо клинических проявлений. Может наблюдаться расширение нисходящего грудного/брюшного отделов аорты, но относительно редко.
3. Глаза. Вывих хрусталика (ectopia lentis) встречается у 60-70% пациентов, но не уникально для данного заболевания. Др. глазные проявления включают раннюю и тяжелую миопию, плоскую роговицу, увеличение осевой длины глазного яблока, гипоплазию радужной оболочки и гипоплазию цилиарной мышцы, что приводит к уменьшению миоза. У пациентов также имеется предрасположенность к отслоению сетчатки, ранней катаракте и глаукоме.
4. Другие системы. При синдроме Марфана увеличивается частота легочных заболеваний: прогрессирующая деформация переднего отдела ГК/грудной сколиоз могут способствовать развитию рестриктивного типа заболевания легких. Кроме того, расширение дистальных отделов ДП у пациентов способствует спонтанному пневмотораксу, который встречается у 15% пациентов. При оценке объема и функции легких следует учитывать избыточный рост длинных костей нижних конечностей, что может привести к снижению нормализованной ФЖЕЛ и общей емкости легких. При коррекции показателей относительно размера ГК/ро-ста в положении сидя результаты исследования функции легких у пациентов с этим заболеванием часто оказываются в норме.
У пациентов с синдромом Марфана кожа обычно имеет нормальную текстуру и эластичность. Наиболее частым признаком со стороны кожных покровов являются стрии — розоватые рубцовые поражения, которые позже становятся белыми (атрофические стрии), возникают у 1/3 пациентов. Они могут возникать при отсутствии ожирения, быстрого набора мышечной массы и беременности, а также на участках, не подверженных чрезмерному растяжению кожи (напр., передняя поверхность плеча/нижняя часть спины). Еще одним частым проявлением является врожденная/приобретенная паховая грыжа. В популяции людей с синдромом Марфана имеет место повышенный риск формирования грыжи после хирургических вмешательств/рецидивирующих грыж.
Расширение дурального мешка/корешковых рукавов (дуральная эктазия) наблюдается у 63-92% пациентов с синдромом Марфана. Хотя эктазия ТМО может привести к появлению боли в поясничном отделе, она часто протекает бессимптомно и диагноз ставится на основании результатов КТ/МРТ пояснично-крестцовой области.
г) Диагностика. Учитывая сложность клинического обследования при синдроме Марфана и соответствующие ДД, диагностику должен координировать профессионал с большим опытом, напр. генетик, кардиолог, офтальмолог. Диагноз основывается на наличии определенного набора ДК, разработанных международной группой экспертов (пересмотренная Гентская нозология синдрома Марфана; табл. 1).
При отсутствии убедительного семейного анамнеза синдрома Марфана диагноз м.б. установлен в 4 разл. ситуациях:
1. Наличие дилатации корня аорты при стандартизации по возрасту и размеру тела (Z-критерий* корня аорты >2)/расслоения аорты в сочетании с эктопией хрусталика позволяет однозначно диагностировать синдром Марфана, независимо от наличия/отсутствия каких-либо системных признаков (см. табл. 1), за исключением случаев, когда они указывают на альтернативный диагноз.
P.S. * Z-критерий показывает, на сколько SD фактический размер аорты превышает должный диаметр аорты (υ — ожидаемое среднее). Z-критерий рассчитывается по следующей формуле: Z = (ФР(Д)А - ДДА)/SE, где ФР(Д)А — фактический размер (диаметр) аорты, ДДА — должный диаметр аорты (υ — ожидаемое среднее по регрессионным моделям), SE — стандартная ошибка среднего, рассчитанная для используемой регрессионной модели. Произвести расчет Z-критерия (Z-score) можно на сайте www.marfan.org/dx/zscore/
2. Наличие дилатации корня аорты (Z-критерий корня аорты >2)/расслоения аорты и выявление достоверной мутации FBN1 (табл. 2) достаточно для установления диагноза, даже при отсутствии эктопии хрусталика.
3. При наличии дилатации корня аорты (Z-критерий корня аорты >2)/расслоения аорты при отсутствии эктопии хрусталика с неизвестным/отрицательным статусом FBN1 диагноз м.б. поставлен на основании присутствия достаточного количества системных признаков (системная оценка >7 баллов; табл. 3). Однако следует исключить признаки, указывающие на альтернативный диагноз, и провести соответствующий альтернативный молекулярный анализ.
4. При наличии эктопии хрусталика, но отсутствии дилатации корня аорты/расслоения аорты для постановки диагноза требуется мутация FBN1, наличие которой ранее приводило к заболеванию аорты. Если мутацию FBN1 однозначно нельзя связать с ССЗ у родственно-го/неродственного пробанда, диагноз пациента следует классифицировать как «синдром изолированной эктопии хрусталика» (см. раздел «Дифференциальный диагноз»).
Несмотря на эти ДК, иногда спорадические случаи у лиц <20 лет могут не соответствовать ни одному из четырех предложенных сценариев, описанных выше. При наличии недостаточного количества системных признаков (системный балл <7) и/или пограничные значения размеров корня аорты (Z-критерий <3) без документально подтвержденных доказательств достоверной мутации FBN1 рекомендуется использовать термин «неспецифическое заболевание соединительной ткани». В случае выявления мутации FBN1 следует использовать термин «потенциальный синдром Марфана».
Человеку с положительным семейным анамнезом синдрома Марфана (если члену семьи был независимо поставлен диагноз с использованием вышеуказанных ДК), заболевание м.б. установлено при наличии:
1) эктопии хрусталика;
2) системной оценки >7 баллов (см. табл. 3);
3) расширения корня аорты с Z-критерием >2 у взрослых (>20 лет) и Z-критерием >3 у лиц <20 лет.
В случае сценариев 2 и 3 признаки, указывающие на альтернативный диагноз, снова должны быть исключены, должен быть проведен соответствующий альтернативный молекулярный анализ.
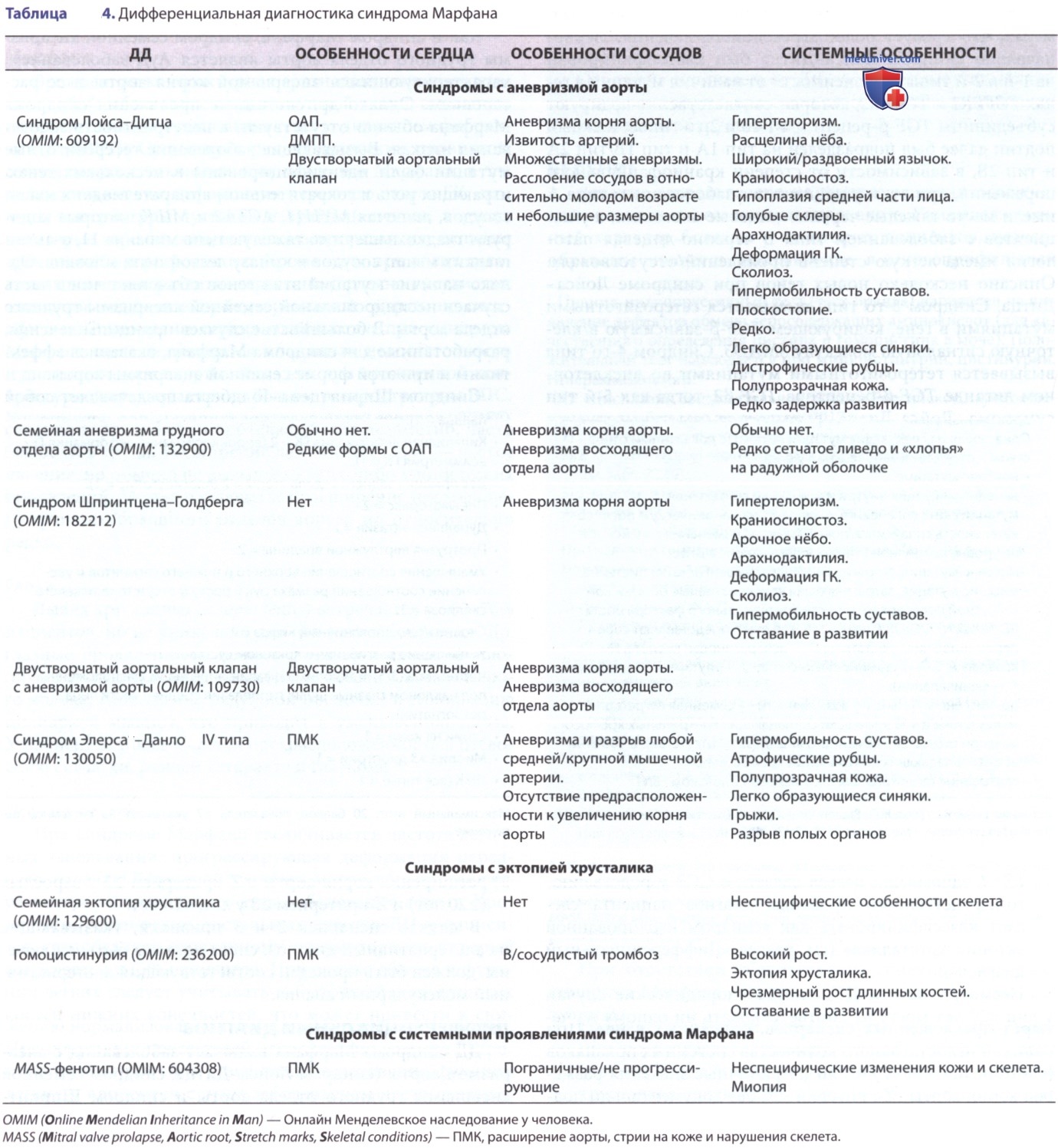
д) Дифференциальный диагноз (ДД). ДД синдрома Марфана включает заболевания с аневризмой аорты (синдром Лойса-Дитца, синдром семейной аневризмы грудного отдела аорты и синдром Шпринтцена-Голдберга), эктопию хрусталика (синдром эктопии хрусталика, синдром Вейля-Маркезани (Weill Georges, Marchesani Oswald) и гомоцистинурия) и системные проявления синдрома Марфана [врожденная контрактурная арахнодактилия и MASS*-фенотип (патологии митрального клапана, аорты, кожи, скелета; (табл. 4))].
P.S. * MASS (Mitral valve prolapse, Aortic root, Stretch marks, Skeletal conditions) — пролапс митрального клапана, расширение аорты, стрии на коже и нарушения скелета.
1. Синдромы с аневризмой аорты. Синдром Лойса-Дитца относится к СЗСТ, характеризуется триадой симптомов: извитость артерий и агрессивное течение аневризмы, гипертелоризм и раздвоение язычка/расщелина нёба, а также множеством черепно-лицевых и скелетных особенностей, характерных для синдрома Марфана. ДД между синдромами Марфана и Лойса-Дитца важна, т.к. в последнем случае характерно расслоение аневризм в более молодом возрасте и при меньших размерах, что требует более агрессивного лечения.
Первоначально синдром Лойса-Дитца был классифицирован на 1-й и 2-й типы, в зависимости от наличия мутации в генах TGFBR1 и TGFBR2, которые соответственно кодируют субъединицы TGF-β-рецептора 1-го и 2-го типов. Каждый подтип далее был подразделен на тип 1А и тип 1В/тип 2А и тип 2В, в зависимости от степени краниофациального поражения, при этом у пациентов с заболеванием типа А имели место тяжелые черепно-лицевые аномалии, а у пациентов с заболеванием типа В черепно-лицевая патология имела легкую степень проявлений/отсутствовала.
Описано несколько новых генов при синдроме Лойса-Дитца. Синдром 3-го типа вызывается гетерозиготными мутациями в гене, кодирующем TGF-β-зависимую в/клеточную сигнальную молекулу SMAD3. Синдром 4-го типа вызывается гетерозиготными мутациями во внеклеточном лиганде TGF-β-рецептора TGF-β2, тогда как 5-й тип синдрома Лойса-Дитца вызывается гетерозиготными мутациями во внеклеточном лиганде TGF-β-рецептора TGF-β3. Типы 3 и 4 характеризуются множественной извитостью артерий, аневризмой и расслоением аорты, а также типичными черепно-лицевыми и скелетными аномалиями. У пациентов со SMAD3-мутациями возможно раннее развитие остеоартрита и наджелудочковых аритмий. В отличие от др. форм синдрома Лойса-Дитца, при 5-м типе не отмечается выраженной извитости артерий и нет убедительных доказательств раннего расслоения аорты.
Как и синдром Марфана, синдром семейной аневризмы грудного отдела аорты является АуД-заболеванием, характеризующимся аневризмой корня аорты и ее расслоением. Однако др. системные проявления синдрома Марфана обычно отсутствуют, а пенетрантность заболевания низкая. Вызывающие заболевание гетерозиготные мутации были идентифицированы в нескольких генах, играющих роль в сократительном аппарате гладких мышц сосудов, включая MYH11, АСТА2 и MILK, которые кодируют гладкомышечную тяжелую цепь миозина 11, α-актин гладких мышц сосудов и киназу легкой цепи миозина. Однако наличие мутаций этих генов объясняет лишь часть случаев несиндромальной семейной аневризмы грудного отдела аорты. В большинстве случаев принципы лечения, разработанные для синдрома Марфана, оказались эффективны и при этой форме семейной аневризмы аорты.
Синдром Шпринтцена-Голдберга представляет собой СЗСТ, которое включает практически все черепно-лицевые, скелетные, кожные и сердечно-сосудистые проявления синдромов Марфана и Лойса-Дитца с дополнительными признаками задержки развития и выраженной гипотонией скелетных мышц. Большинство случаев вызвано гетерозиготными мутациями в гене SKI, который кодирует в/клеточный репрессор передачи сигналов TGF-β. Вовлечение сосудов имеет тенденцию быть менее распространенным и менее тяжелым по сравнению с синдромами Марфана и Лойса-Дитца.
2. Синдромы с эктопией хрусталика. И синдром эктопии хрусталика, и синдром Вейля-Маркезани также м.б. вызваны гетерозиготными мутациями в FBN1. Недавно было показано, что сложные гетерозиготные/гомозиготные мутации во втором локусе ADAMTSL4 вызывают синдром эктопии хрусталика с несколько более молодым возрастом на момент постановки диагноза. Интересно, что некоторые мутации FBN1 могут вызывать классический синдром Марфана, синдром эктопии хрусталика и эктопию хрусталика в сочетании с кожными, но не сердечно-сосудистыми проявлениями синдрома Марфана, это позволяет предположить, что данные проявления являются частью спектра клинических признаков одного и того же заболевания, и подчеркивает потенциальный вклад генетических модификаторов заболевания.
Синдром Вейля-Маркезани является СЗСТ, характеризующимся аномалиями кожи, скелета и глаз, включая микросферофакию, эктопию хрусталика и миопию. Признаки, исключающие диагноз синдрома Марфана, включают низкий рост и брахидактилию. В дополнение к мутациям FBN1 (тип 2) синдром м.б. вызван гомозиготными/сложными гетерозиготными мутациями в ADAMTS10 (тип 1)/гомозиготными мутациями в LTBP2 (тип 3), которые кодируют металлопептидазу ADAM с тромбоспондиновым мотивом 10 типа 1 и латентный TGF-β1, связывающий белок 2, соответственно.
Гомоцистинурия — нарушение обмена в-в, вызванное гомозиготными/сложными гетерозиготными мутациями в гене, кодирующем цистатионин-β-синтазу, что приводит к повышению уровня как гомоцистеина, так и метионина. Клинические признаки нелеченной гомоцистинурии включают эктопию хрусталика и скелетные аномалии, напоминающие синдром Марфана. Однако, в отличие от последнего, больные часто страдают задержкой развития, предрасположенностью к тромбоэмболическим явлениям и высокой частотой ИБС.
3. Синдромы с системным проявлением синдрома Марфана. Врожденная контрактурная арахнодактилия представляет собой заболевание соединительной ткани, вызванное гетерозиготными мутациями в гене, кодирующем фибриллин-2 (FBN2). Существует ряд клинических признаков, совпадающих с таковыми при синдроме Марфана, в т.ч. долихостеномелия, деформация переднего отдела ГК, сколиоз, контрактуры суставов и арахнодактилия, а также некоторые черепно-лицевые аномалии развития, включая арочное нёбо и ретрогнатию.
Кроме того, при обеих патологиях возможно развитие серьезных сердечно-сосудистых аномалий, ведущих к преждевременной смерти, но специфические аномалии сердца отличаются; при синдроме Марфана чаще встречаются клапанная недостаточность и дилатация корня аорты, тогда как при врожденной контрактурной арахнодактилии больше распространены ВПС. У пациентов с врожденной контрактурной арахнодактилией имеет место измененная форма ушных раковин — «мятое» ухо (отличительный признак этого состояния).
У многих пациентов, направленных по поводу возможного синдрома Марфана, обнаруживаются признаки СЗСТ, включая длинные конечности, деформацию ГК, атрофические стрии, ПМК и пограничную, но не прогрессирующую дилатацию корня аорты, которые при этом не соответствуют ДК синдрома Марфана. Такое сочетание признаков обозначается аббревиатурой MASS-фенотип, подчеркивая митральные, аортальные, кожные и скелетные аномалии. MASS-фенотип может сегрегировать на большие родословные признаки и оставаться стабильным с течением времени. Диагноз особенно сложен в контексте молодого пациента со спорадическим заболеванием, за которым необходимо тщательное наблюдение с целью ДД MASS-фенотипа и формирующегося синдрома Марфана.
Семейный синдром ПМК также м.б. вызван мутациями в гене, кодирующем фибриллин-1, и включать субдиагностические системные проявления.
е) Лабораторные данные. Для исключения дефицита цистатионин-β-синтазы (гомоцистинурии) лабораторно необходимо подтвердить «-» результат цианид-нитропруссидного теста* мочи/анализа на специфические аминокислоты. Хотя считается, что большинство, если не все, люди с классическим синдромом Марфана имеют мутацию FBN1, большой размер этого гена и крайняя аллельная гетерогенность синдрома не позволяют провести эффективную молекулярную диагностику.
P.S. * Цианид-нитропруссидный тест (тест Бранда) направлен на выявление дефектов обмена серосодержащих аминокислот (для качественного определения цистина и гомоцистина в моче). Положительный тест наблюдается при гомоцистинурии, цистинурии, гипераммониемии.
Результаты скрининга мутаций зависят от техники и клинической картины. Остается неясным, являются ли «отсутствующие» мутации просто нетипичными по характеру/локализации в пределах FBN1 или находятся в др. гене. Др. альтернативные заболевания, такие как MASS-фенотип, эктопия хрусталика, синдром Вейля-Маркезани и синдром Шпринтцена-Голдберга, сопряжены с мутациями в гене FBN1. Часто бывает сложно/невозможно предсказать фенотип по характеру/локализации мутации FBN1 при синдроме Марфана. Следовательно, молекулярно-генетические методы могут способствовать диагностике, но они не заменяют комплексную клиническую оценку и последующее наблюдение. Отсутствие/наличие мутации FBN1 недостаточно для исключения/подтверждения диагноза соответственно.
Основные молекулярно-генетические критерии и маркеры рисков формирования клинических проявлений синдрома Марфана:
• Ген и хромосомная локализация — CoLla2-17q21.3-q22, >100 мутаций.
• Основные клинические ДК — скелетные аномалии, аневризма аорты, пролапсы створок клапанов, вывихи и подвывихи хрусталика**.
P.S. ** КР «Дисплазии соединительной ткани», Российское научное медицинское общество терапевтов, 2017 г.
ж) Ведение. Лечение сосредоточено на предотвращении осложнений и генетическом консультировании. Учитывая сложный характер болезни у некоторых пациентов, целесообразным является направление в многопрофильный центр, где генетик, имеющий опыт работы с синдромом Марфана, работает совместно со специалистами узкого профиля для координации и рационального подхода к мониторингу и лечению. Ежегодное обследование на предмет ССЗ, сколиоза и офтальмологических проблем является обязательным.
з) Современные методы терапии. Большинство доступных в настоящее время/находящихся на стадии изучения методов лечения направлены на нивелирование сердечно-сосудистых осложнений и включают ограничение активности, операции на аорте, профилактику эндокардита и современные фармакологические подходы.
1. Ограничение активности. Физиотерапия способствует улучшению работы ССС, нервно-мышечного тонуса и психосоциального здоровья, поэтому рекомендуется умеренная аэробная нагрузка. Однако следует избегать интенсивных ФН, соревновательных/контактных видов спорта, и особенно изометрических упражнений, таких как поднятие тяжестей, которые требуют выполнения маневра Вальсальвы (Valsalva maneuver).
2. Операции на аорте. Исход хирургического вмешательства более благоприятен, если оно проводится в плановом, а не в срочном/экс-тренном порядке (смертность 1,5% против 2,6% и 11,7% соответственно). Поэтому хирургическое вмешательство на аорте следует рекомендовать взрослым пациентам при диаметре корня аорты ~50 мм, при этом раннее вмешательство следует рассматривать в отношении пациентов с быстрым прогрессированием [>5-10 мм/год (>2 мм/год**)]/ семейным анамнезом раннего расслоения аорты. Не существует четких критериев, определяющих время проведения операции у детей, у которых расслоение аорты встречается крайне редко, независимо от ее размеров.
Это побудило многие медцентры принять критерий для взрослого населения 50 мм, хотя раннее хирургическое вмешательство м.б. предпринято при высокой скорости прогрессирования (>10 мм/год)/появлении значительной аортальной регургитации. Сохранение нативного аортального клапана во время операции рекомендуется во избежание необходимости пожизненной АКТ. Восстановление/замена митрального клапана рекомендуется при тяжелой регургитации митрального клапана с сопутствующими симптомами/ прогрессирующей дилатацией и дисфункцией ЛЖ.
{2С} Показания к операции при дилатации аорты наиболее точно разработаны для синдрома Марфана: дилатация аорты >5 см; в случае меньших размеров (>45 мм) — при наличии факторов риска (семейный анамнез диссекции аорты, увеличение диаметра расширение >2 мм/год, выраженная аортальная регургитация, планируемая беременность)**.
P.S. ** КР «Дисплазии соединительной ткани», Российское научное медицинское общество терапевтов, 2017 г.
3. Беременность. У женщин с синдромом Марфана существует более высокий риск расслоения аорты во время беременности. Однако лучшее понимание и данные о заболевании показали, что риск у пациентов с диаметром корня аорты <40 мм невелик. Профилактическое протезирование корня аорты может минимизировать риск расслоения аорты и смерти у женщин с синдромом Марфана, желающих забеременеть, но это вмешательство не влияет на риск расслоения более дистальных отделов восходящей/нисходящей аорты.
4. Профилактика эндокардита. Профессиональный консультативный совет Национального фонда Марфана считает, что пациентам с данным заболеванием должна длительно проводиться профилактика бактериального эндокардита, отчасти в связи с точно не установленной, но вероятной возможностью предпочтительного поражения бактериальным агентом миксоматозных клапанов, типичных для синдрома Марфана.
5. Современные фармакологические подходы. β-Блокаторы традиционно считались стандартом лечения синдрома Марфана, и многочисленные небольшие наблюдательные исследования показали их защитный эффект на рост корня аорты, при этом доза обычно титруется до достижения ЧСС покоя <100 ударов/мин при субмаксимальной ФН. С учетом предполагаемой роли гемодинамического стресса в дилатации и расслоении аорты при синдроме Марфана эти эффекты объясняются отрицательными инотропным и хронотропным эффектами β-блокады.
и) Новые терапевтические стратегии:
1. Блокаторы рецепторов ангиотензина II типа 1. Существуют обширные доказательства связи передачи сигналов ангиотензином II с активацией и передачей сигналов TGF-β. На мышиной модели синдрома Марфана было показано, что блокатор рецепторов ангиотензина II типа 1 лозартан полностью предотвращает патологический рост корня аорты и нормализует как толщину, так и структуру стенки аорты — эффекты, которые отсутствовали у мышей из группы плацебо и пропранолола. Эти данные указывают на возможность продуктивного ремоделирования стенки аорты при синдроме Марфана после ингибирования TGF-β. Примечательно, что при применении лозартана у этих мышей также отмечалось уменьшение патологических проявлений со стороны легких и скелетных мышц, а это еще раз подтверждает, что лечение основано на ингибировании передачи сигналов TGF-β, а не на просто снижении гемодинамической нагрузки на структурно предрасположенные ткани.
В подтверждение значимости для человека ретроспективное исследование, оценивающее эффект блокаторов рецепторов ангиотензина у небольшой когорты педиатрических пациентов с синдромом Марфана с выраженным расширением корня аорты, несмотря на предыдущую альтернативную медикаментозную терапию, показало, что данные ЛП значительно замедляют скорость роста корня аорты и синотубулярного соединения (оба явления характерны для синдрома Марфана), тогда как дистальный отдел восходящей аорты (который обычно не расширяется при синдроме Марфана) остался неизменным. Дополнительные доказательства положительного эффекта от терапии лозартаном были предоставлены в трех проспективных клинических испытаниях, демонстрирующих, что лечение [лозартан ± p-блокатор] замедляло прогрессирование роста корня аорты у пациентов с синдромом Марфана.
Сравнительное клиническое исследование, оценивающее терапевтический эффект лозартана по сравнению с атенололом у пациентов с синдромом Марфана, установило, что оба ЛП обеспечивают достаточную защиту от дилатации аорты без существенной разницы в терапевтическом эффекте между этими ЛП, несмотря на использование стандартной дозы лозартана (одобренная FDA доза для АГ) и обычно высокой дозы атенолола (СрСД атенолола в 1,5 раза, а максСД — в 2 раза превышали верхний предел, одобренный FDA для лечения АГ). Обе группы в этом исследовании показали очень медленную скорость роста корня аорты и значительное снижение Z-критерия корня аорты с течением времени, что превосходило показатели, наблюдаемые у нелеченых пациентов с синдромом Марфана/пациентов, получавших стандартную дозу атенолола (~ 1 мг/кг/сут). Эти данные убедительно свидетельствуют в пользу наличия терапевтического потенциала обоих методов у пациентов с синдромом Марфана.
2. Ингибиторы ангиотензин-превращающего фермента. Было высказано предположение, что иАПФ могут оказаться также/более эффективными, чем блокаторы рецепторов ангиотензина, при лечении синдрома Марфана благодаря их способности ограничивать передачу сигналов через рецепторы ангиотензина как 1-го, так и 2-го типов. Однако эксперименты на мышах показали, что положительный эффект лозартана опосредуется как блокированием передачи сигналов рецептора ангиотензина 1-го типа, так и отклонением передачи сигналов через рецептор ангиотензина 2-го типа, и, как следствие, эналаприл, хотя и превосходит плацебо, оказался менее эффективным, чем лозартан. Эти результаты согласуются с рядом небольших исследований, предполагающих некоторую защиту, обеспечиваемую иАПФ, у пациентов с синдромом Марфана, однако для решения этой проблемы необходимы дополнительные эксперименты и клинический опыт.
3. Ингибиторы киназы, регулируемой внеклеточными сигналами. Известно, что лиганд-зависимая активация рецептора TGF-β может инициировать неканонические каскады, включая МАРК*. В аорте мышей с синдромом Марфана наблюдается усиление активации киназ, регулируемых внеклеточными сигналами 1-го и 2-го типов, в то время как введение внутрь биодоступного селективного ингибитора активации киназ, регулируемых внеклеточными сигналами 1-го и 2-го типов, — RDEA119 полностью предотвращает патологическое расширение корня аорты у этих мышей. Это позволяет предположить, что киназы, регулируемые внеклеточными сигналами 1-го и 2-го типов, являются критическими медиаторами патофизиологии заболевания и потенциальной жизнеспособной терапевтической мишенью.
P.S. * МАРК (Mitogen-Activated Protein Kinase) — митоген-активируемая протеинкиназа.
к) Прогноз. Основной причиной смертности является дилатация, расслоение и разрыв корня аорты, при этом большинство смертельных исходов приходится на 3-е и 4-е десятилетия жизни. Повторная оценка ОПЖ при синдроме Марфана показала, что ранняя диагностика и усовершенствованное медикаментозное и хирургическое лечение значительно улучшили прогноз для пациентов с этим заболеванием. Тем не менее синдром Марфана по-прежнему сопряжен со значительной заболеваемостью, а в отдельных подгруппах отмечаются невосприимчивость к терапии и ранняя смертность. В обзоре 54 пациентов с установленным в младенчестве диагнозом 89% имели серьезную патологию сердца, которая прогрессировала, несмотря на стандартную терапию (22% умерли в детском возрасте, 16% — в возрасте <1 года).
По оценкам, при классической форме синдрома Марфана >90% людей будут иметь ССЗ в течение жизни, что вызывает как физический, так и психический стресс у пациентов и их семей. Осведомленность об этих проблемах и направление в службы поддержки могут способствовать формированию позитивного взгляда на это состояние.
л) Генетическое консультирование. Наследственный характер синдрома Марфана делает обязательным консультирование по поводу риска (генетического) рецидива. Возраст отцов в спорадических случаях в среднем на 7-10 лет больше, чем возраст отцов в общей популяции. Этот эффект отцовского возраста предполагает, что данные случаи представляют собой новые доминантные мутации с минимальным риском рецидива для будущего потомства здоровых родителей. Из-за редких сообщений о гонадном мозаицизме у фенотипически нормальных родителей риск рецидива для родителей в случае спорадического заболевания можно считать низким, но не нулевым. Однако каждый ребенок больного родителя имеет 50% риск унаследовать мутацию синдрома Марфана и, т.о., заболеть. Консультации по вопросам риска рецидива лучше всего проводить профессионалу, обладающему опытом в вопросах, связанных с синдромом Марфана.