1. Доброкачественные опухоли щитовидной железы. Доброкачественные опухоли ЩЖ составляют 75% от всех новообразований ЩЖ детского возраста. Исследование подозрительного узла ЩЖ включает лабораторную оценку функции ЩЖ, УЗИ для определения характеристик узла (узлов) ЩЖ и регионарных ЛУ, а также ТИАБ под контролем УЗИ в качестве метода цитопатологической диагностики. Ядерно-нуклидная сцинтиграфия с использованием радиоактивного изотопа йода (123I) или технеция-99m (в виде натрия пертехнетата [99mTc) не рекомендуется при первоначальной диагностике, за исключением случаев сниженного уровня ТТГ.
2. Злокачественные опухоли щитовидной железы. Злокачественные опухоли ЩЖ редки в детском возрасте. Тем не менее, у детей могут развиваться медуллярная карцинома ЩЖ (МКЩЖ) и дифференцированные карциномы ЩЖ (ДКЩЖ), которые, в свою очередь, представлены папиллярной (ПКЩЖ) и фолликулярной карциномами ЩЖ. ПКЩЖ составляет подавляющее большинство случаев рака ЩЖ у детей. Заболеваемость раком ЩЖ ↑ с пиком заболеваемости в 15-19 лет. За редким исключением, дети с карциномой ЩЖ имеют отличный прогноз, с ожидаемой выживаемостью в несколько десятилетий, даже при наличии метастатического распространения на момент постановки диагноза.
Основным точно установленным фактором риска развития ПКЩЖ является воздействие ионизирующего излучения.
МКЩЖ редко развивается в детском возрасте, почти всегда возникает как часть наследственного АуД-синдрома эндокринных опухолевых заболеваний, возникающего вторично по отношению к активирующим мутациям в протоонкогене RET (REarranged during Transfection), известного как множественная эндокринная неоплазия типа 2А или 2В. При самых распространенных мутациях в протоонкогене RET наблюдается практически полная пенетрантность МКЩЖ. Кроме того, множественная эндокринная неоплазия типа 2А или 2В также характеризуется высоким риском развития феохромоцитомы (до 50% в течение жизни).
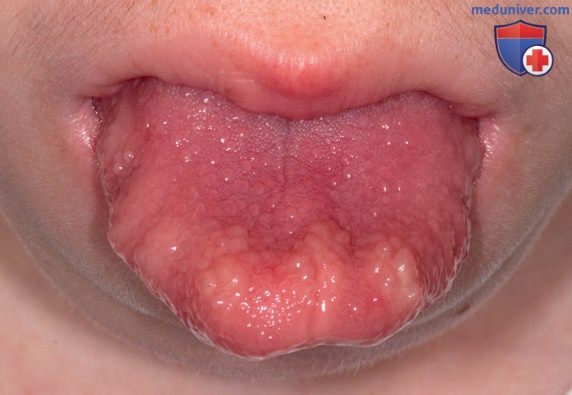
У 20% пациентов с множественной эндокринной неоплазией типа 2А также развивается первичный гиперпаратиреоз. У пациентов с множественной эндокринной неоплазией типа 2В гиперпаратиреоз не развивается, но наблюдается выраженный клинический фенотип, включающий характерную внешность, марфаноидный тип телосложения, ганглионейроматоз ЖКТ и невромы слизистой оболочки полости рта (рис. 1). Диагноз множественной эндокринной неоплазии типа 2В нередко устанавливается на поздней стадии (обычно уже после метастазирования МКЩЖ), поскольку его патогномоничные признаки с трудом распознаются в раннем детстве, хотя самыми ранними характерными проявлениями заболевания являются отсутствие слез при плаче и запоры.
Рисунок 1. Классический внешний вид невромы слизистой оболочки полости рта на языке у мальчика с множественной эндокринной неоплазией типа 2В, вторичной по отношению к типичной мутации М918Т в протоонкогене RET
МКЩЖ м.б. спорадической или семейной без признаков множественной эндокринной неоплазии типа 2А или 2В. Кроме того, МКЩЖ может сочетаться с болезнью Гиршпрунга.
Рак ЩЖ у детей обычно проявляется в виде бессимптомного образования ЩЖ и/или шейной лимфаденопатией, хотя детям с множественной эндокринной неоплазией типа 2 диагноз часто ставится только после «+» результата генетического исследования, а пациентам с множественной эндокринной неоплазией 2В — после распознавания клинического фенотипа. В большинстве случаев ПКЩЖ наблюдаются метастазы в ЛУ, при этом метастазы в легких обнаруживаются у 20% пациентов, в основном у детей с массивным поражением ЛУ. МКЩЖ также часто метастазирует в шейные ЛУ
Первичной терапией рака ЩЖ является тотальная тиреоидэктомия и, по показаниям, селективная лимфо-диссекция, выполняемая опытным хирургом, специализирующимся на раке ЩЖ. При ДКЩЖ радиоактивный изотоп йода (131I) используется после хирургической резекции для лечения отдаленных метастазов, накапливающих йод, и нерезектабельной остаточной опухоли шеи. При ПКЩЖ 131I применяется только для лечения детей из группы высокого риска, для которых такое лечение может оказаться эффективным.
Дети с МКЩЖ не нуждаются в терапии 131I. При ДКЩЖ уровень ТТГ изначально подавляется введением супрафизиол. дозы левотироксина натрия, поскольку ТТГ может стимулировать рост ДКЩЖ, а при МКЩЖ уровень ТТГ остается в норме. FDA одобрило пероральные ингибиторы тирозинкиназы для лечения поздних стадий МКЩЖ и ДКЩЖ у взрослых, но педиатрические пациенты редко нуждаются в такой терапии. Долгосрочное наблюдение включает мониторинг уровней опухолевых маркеров (тиреоглобулин/ АТл к тиреоглобулину при ДКЩЖ, кальцитонин/раковый эмбриональный АГн при МКЩЖ), а также регулярные визуализирующие исследования, в первую очередь УЗИ шеи.
Были выявлены и детально задокументированы корреляции между генотипом и фенотипом при множественной эндокринной неоплазии 2, а биологическая агрессивность МКЩЖ зависит от особенностей наследственности. С появлением генетического тестирования на наличие мутаций в протоонкогене RET МКЩЖ стала одной из немногих ЗНО, которые можно вылечить с помощью ранней тиреоидэктомии до того, как опухоль начнет метастазировать. Рекомендации относительно возраста, подходящего для хирургического лечения детей, являющихся носителями мутации в протоонкогене RET, постоянно видоизменяются и дорабатываются, но предписывают проводить перед операцией клинические исследования, особенно на определение уровня кальцитонина и распознавание генотипа, кроме того, следует учитывать предпочтения родителей пациента.
б) Рак носоглотки. Рак носоглотки встречается у детей довольно редко, но вместе с тем это одно из самых распространенных ЗНО носоглотки у педиатрических пациентов. Среди взрослых самая высокая заболеваемость отмечается в Южном Китае, кроме того, высокие уровни заболеваемости наблюдаются среди инуитов, а также в Северной Африке и Северо-Восточной Индии. В Китае рак носоглотки редко развивается в детском возрасте, но в др. странах значительная часть случаев приходится на детскую возрастную группу, в первую очередь на подростков. У мальчиков эта опухоль развивается в два раза чаще, чем у девочек, и > встречается у афроамериканцев. У детей рак носоглотки обычно имеет недифференцированную гист. структуру и ассоциируется с ВЭБ.
Рак носоглотки связан со специфическими типами HLA, но и др. генетические факторы могут играть роль, особенно в популяциях с низкой заболеваемостью.
У большинства пациентов детского возраста наблюдается распространенное местно-регионарное заболевание, проявляющееся как шейная лимфаденопатия. Также могут присутствовать носовые кровотечения, тризм жевательной мускулатуры и поражение ЧМН. Диагноз устанавливается на основании биопсии носоглотки или шейных ЛУ. В большинстве случаев уровень ЛДГ повышен, но этот показатель не является специфическим. Для оценки распространенности местно-регионарного заболевания проводят КТ или МРТ головы и шеи. Для исключения метастазов применяется РОГК, КТ, сцинтиграфия костей скелета и печени. ПЭТ является, по-видимому, эффективным методом мониторинга первичного заболевания и выявления метастазов.
Уровни ДНК ВЭБ коррелируют со стадией заболевания, имеют прогностическое значение и могут использоваться для мониторинга рецидивов.
РФ: всем пациентам с подозрением на рак носоглотки определяют АТл к капсидному АГн ВЭБ, IgG к ранним белкам ВЭБ, IgG к ядерному АГн ВЭБ в крови, чтобы оценить активность вирусной инфекции. Также проводят эпифарингоскопию для уточнения распространения опухоли на своды носоглотки*.
P.S. * Рак носоглотки, 2020 г. (Российское общество детских онкологов; Национальное общество детских гематологов и онкологов.)
Стандартное лечение представляет собой комбинацию лучевой терапии и XT на основе цисплатина, который вводится до или одновременно с облучением с или без неоадъювантной XT.
РФ: пациентам с установленным диагнозом «рак носоглотки» и отсутствием эффекта со стороны метастазов в регионарных ЛУ (по данным обследования) рекомендуется хирургическое лечение — двусторонняя лимфодиссекция после завершения консолидирующего этапа химиолучевой терапии*.
P.S. * Рак носоглотки, 2020 г. (Российское общество детских онкологов; Национальное общество детских гематологов и онкологов.)
Исход зависит от степени распространенности опухоли. У пациентов с отдаленными метастазами прогноз очень плохой. Применение лучевой терапии с модуляцией интенсивности улучшает местный контроль и ↓ риск развития побочных эффектов, связанных с лучевой терапией, таких как гормональная дисфункция, кариес, фиброз и вторичные ЗНО. Применение протонной терапии может привести к дальнейшему уменьшению побочных эффектов.
в) Рак толстой и прямой кишки. Колоректальная карцинома редко развивается в детском возрасте, уровень заболеваемости колоректальной карциномой составляет ~1:1 000 000. Даже у пациентов с предрасполагающими заболеваниями колоректальная карцинома обычно не проявляется до позднего подросткового или зрелого возраста. Наследственный неполипозный рак толстой кишки является АуД-заболеванием, при котором герминальные мутации в генах, ответственных за систему коррекции неспаренных оснований ДНК, вызывают ошибки в репарации ДНК и микросателлитную нестабильность.
Семейный аденоматозный полипоз и аттенуированная (слабовыраженная) форма семейного аденоматозного полипоза являются аутосомными заболеваниями с герминальными мутациями в гене АРС.
Так же, как и при колоректальной карциноме, пациенты с наследственным неполипозным раком толстой кишки и аттенуированной формой семейного аденоматозного полипоза имеют предрасположенность к ряду некишечных онкологических заболеваний. Десмоидные опухоли могут развиваться у пациентов с семейным аденоматозным полипозом, в то время как у пациентов с наследственным неполипозным раком толстой кишки повышен риск развития опухолей МПС, желудка и тонкой кишки. MYH-ассоциированный полипоз, синдром Пейтца-Егерса и ювенильный полипоз также предрасполагают к колоректальной карциноме.
Генетический анализ и скрининг на онкологические заболевания пациентов с наследственным неполипозным раком толстой кишки и семейным аденоматозным полипозом следует начинать в детском или подростковом возрасте. Аналогичным образом, генетическое обследование на наличие этих заболеваний следует проводить у пациентов детского и подросткового возраста с раком толстой кишки, даже если в анамнезе нет предрасполагающих генетических факторов.
Характерными признаками колоректальной карциномы являются стул с кровью или мелена, боль в животе, снижение МТ и нарушения работы кишечника. Во многих случаях симптомы слабо выражены, поэтому диагноз часто ставят на поздних стадиях заболевания. Гист. структура колоректальной карциномы детского возраста отличается от таковой у взрослых: большинство детских опухолей представляют собой либо муцинозную аденокарциному, либо перстневидно-клеточную карциному. У детей колоректальная карцинома, как правило, проявляется на более поздних стадиях. Лечение заключается в хирургической резекции, когда это возможно, при нерезектабельных опухолях назначается XT.
Во время хирургической резекции первичной опухоли д.б. проведено адекватное удаление ЛУ В некоторых случаях эффективной м.б. и лучевая терапия. У педиатрических пациентов общий прогноз хуже, чем у взрослых, но причины этого различия неизвестны.
г) Рак надпочечников. Адренокортикальные опухоли развиваются из коркового слоя надпочечников, тогда как феохромоцитомы исходят из хромаффинных клеток мозгового в-ва надпочечников, вырабатывающих катехоламины. Опухоли, возникающие из парасимпатических и симпатических параганглиев, находящихся вне мозгового в-ва надпочечников, называются параганглиомами. Гистопатологическая классификация опухолей надпочечников как «доброкачественных» или «злокачественных» не всегда хорошо коррелирует с клинической картиной, что затрудняет дифференцирование ЗНО от доброкачественных процессов на основании данных только гистопатологических исследований. Поэтому необходимо длительное наблюдение.
Из-за тесной связи этих опухолей с генетическими заболеваниями всем пациентам с диагнозом адренокортикальной опухоли или феохромоцитомы/параганглиомы рекомендуется пройти генетическое консультирование.
Адренокортикальные опухоли встречаются очень редко и, как правило, проявляются в возрасте <10 лет. Девочки болеют > мальчиков. Более чем в 90% случаев такие опухоли являются функциональными, в основном они продуцируют андрогены и вызывают клинически выраженную вирилизацию, хотя может также наблюдаться гиперсекреция кортизола. Адренокортикальные опухоли также могут проявляться в виде объемного образования в БП или болевым синдромом.
У детей адренокортикальные опухоли чаще всего связаны с синдромом Ли-Фраумени (герминальные инактивирующие мутации в гене-супрессоре опухоли ТР53) и синдромом Беквита-Видемана, но они также могут наблюдаться при гемигиперплазии, отличной от синдрома Беквита-Видемана, множественной эндокринной неоплазии 1-го типа, синдроме Маккуна-Олбрайта, семейном аденоматозном полипозе и очень редко — при врожденной гиперплазии надпочечников. Нетипичные причины двустороннего узлового заболевания коры надпочечников, которое обычно сопровождается синдромом Кушинга, включают Карни-комплекс и макронодулярную гиперплазию коры надпочечников.
Феохромоцитомы/параганглиомы — это редкие опухоли, которые у детей чаще всего бывают двусторонними, злокачественными и вторичными по отношению к наследственному опухолевому синдрому. Существует также тесная связь между феохромоцитомами/параган-глиомами и ВПС «синего типа». У детей феохромоцитомы/параганглиомы чаще всего сочетаются с такими генетическими нарушениями, как болезнь Гиппеля-Линдау и семейными синдромами параганглиомы (1, 2, 3, 4 типов), вызванными мутациями в гене сукцинатдегидрогеназы.
Кроме того, необходима ДД параганглиомы с множественной эндокринной неоплазией 2 (типы 2А и 2В) и нейрофиброматозом 1-го типа, хотя эти заболевания в большей степени ассоциируются с параганглиомами, развивающимися у взрослых. У детей с феохромоцитомами/параганглиомами обычно наблюдается устойчивая гипертензия, при этом классическая триада признаков (головная боль, сердцебиение и диафорез), наблюдаемая у взрослых, у детей может отсутствовать. СДВГ также распространен среди детей с этими опухолями. Лучшим скрининговым тестом на выявление феохромоцитомы/параганглиомы является измерение уровня метанефрина в плазме крови и/или моче.
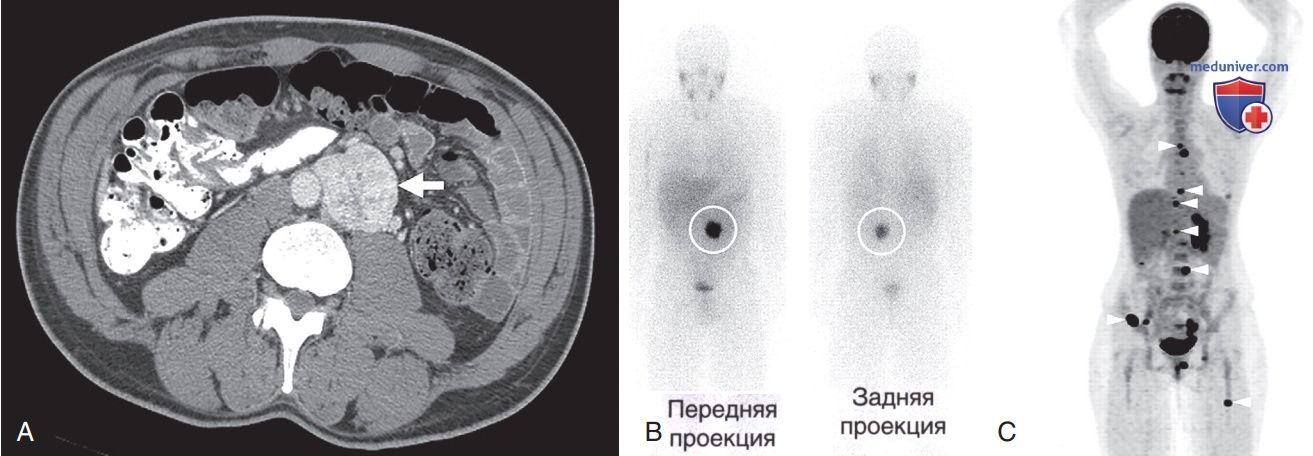
Визуализирующие исследования включают КТ, МРТ и ПЭТ с метайодбензилгуанидином или более чувствительным методом — ПЭТ-сканированием (рис. 2).
Рисунок 2. Параганглиома: А — постконтрастное компьютерно-томографическое изображение в аксиальной проекции параганглиомы у пациента с признаками избытка катехоламинов и норадренергическим биохимическим профилем. Параганглиома (указано стрелкой) локализована в парааортальном пространстве между левой почечной артерией и началом нижней брыжеечной артерии в области аортальных параганглиев; В — сцинтиграфия с метайрдбензилгуанидином, у того же пациента через 24 ч после введения 123I-метайрдбензилгуанидина, демонстрирующее интенсивное накопление радиофармпрепарата в опухоли; C — позитронно-эмиссионная томография с флудезоксиглюкозой [18F] («Фтордезоксиглюкозой, 18F») у пациента с метастатической формой параганглиомы и мутацией в гене SDHB. Преобладают метастазы в костях (указано короткими стрелками)
Первичное лечение адренокортикальной опухоли и феохромоцитомы/параганглиомы включает хирургическое вмешательство, проводимое опытным хирургом. Дети с феохромоцитомой/параганглиомой нуждаются в предоперационном медикаментозном лечении с применением α- и β-адреноблокаторов. Терапия первой линии метастатической формы адренокортикальной опухоли включает митотан и XT ЛП цисплатина, этопозида и доксорубицина. Метастатические формы феохромоцитомы/ параганглиомы ранее лечились циклофосфамидом, вин-кристином и дакарбазином.
В настоящее время изучаются новые ЛП направленного действия для лечения прогрессирующих метастатических форм адренокортикальной опухоли и феохромоцитомы/параганглиомы, которые обычно не реагируют на стандартные XT-схемы. Для облегчения симптомов и улучшения качества жизни показана эндокринная терапия, мишенью которой служит гормональная гиперпродукция для облегчения симптомов и улучшения качества жизни.
д) Десмопластическая мелкокруглоклеточная опухоль. Десмопластическая мелкокруглоклеточная опухоль — очень редкая и агрессивная опухоль мезенхимальной природы, возникающая преимущественно у подростков и молодых взрослых мужчин. Характерным признаком десмопластической мелкокруглоклеточной опухоли является диагностическая хромосомная транслокация между геном опухоли Юинга и геном опухоли Вильмса, t(11;22)(p13;q12), в результате которой появляется химерный ген (EWS-WT1), кодирующий химерный белок с новыми онкогенными свойствами.
Десмопластическая мелкокруглоклеточная опухоль обычно проявляется на поздней стадии в виде объемного образования в БП, множественными метастазами в забрюшинном пространстве и области сальника, а также признаками саркоматоза БП, включающими боль, асцит, кишечную непроходимость, гидронефроз и снижение МТ. Десмопластическая мелкокруглоклеточная опухоль развивается преимущественно в БП, но может распространяться в ЛУ, печень, легкие и кости. Стандартной терапии на сегодняшний день не установлено. Агрессивное лечение с использованием комбинированной XT, циторедуктивной хирургии и облучения всей БП почти во всех случаях → плохой исход.
Медиана выживаемости составляет от 17 до 25 мес, а 5-летняя общая выживаемость не >20%. Хотя высокодозированная XT с последующей трансплантацией аутологичных стволовых клеток оказывает некоторый благоприятный эффект, от этого метода отказались из-за его высокой токсичности. В настоящее время изучаются альтернативные варианты лечения, включая гипертермическую в/брюшинную XT и радиоиммунотерапию с использованием моноклональных АТл, направленных на разл. поверхностные АГн опухолевых клеток.