Гистиоцитозы детского возраста представляют собой разнообразную группу расстройств с тяжелыми клиническими проявлениями. Эти заболевания редко встречаются изолированно, а скорее группами, т.к. они все характеризуются значительной пролиферацией или накоплением клеток моноцитарно-макрофагальной системы, происходящих из костного мозга (миелоидного происхождения). Несмотря на похожесть клинической картины этих заболеваний, в каждом случае необходим точный ДД для выбора наиболее оптимального метода лечения.
В настоящее время разработана систематическая классификация гистиоцитозов, которая основана на данных гистопатологических исследований (табл. 1). Т.о., при подозрении на гистиоцитоз необходимо провести тщательный анализ биопсийных образцов, полученных в ходе диагностики. Анализ биоптата проводится с помощью таких методов, как иммуноокрашивание, молекулярный анализ и электронная микроскопия. В некоторых случаях может потребоваться специальная обработка биопсийного образца.
а) Гистопатологическая классификация. На основании данных гистопатологических исследований выделяют 3 класса гистиоцитозов детского возраста. Наиболее известным является гистиоцитоз из клеток Лангерганса, ранее называвшийся гистиоцитозом X. Гистиоцитоз из клеток Лангерганса включает такие нарушения, как ограниченные поражения костей или кожи, эозинофильную гранулему, болезнь Хенда-Шюллера-Крисчена* и болезнь Леттерера-Сиве**.
P.S. * Болезнь Хенда-Шюллера-Крисчена или липидный гранулематоз — это генерализованная форма гистиоцитоза X. Характеризуется хроническим волнообразным течением.
P.S. ** Болезнь Абта-Леттерера-Сиве — острая распространенная форма гистиоцитоза X, характеризующаяся образованием очагов разрастания атипических гистиоцитов в коже, костях, внутренних органах. Клиническая картина заболевания впервые была описана в 1924 г. Леттерером.
В норме клетки Лангерганса являются АГн-презентирующими клетками кожи. Отличительной особенностью всех форм гистиоцитоза из клеток Лангерганса является клональная пролиферация дендритных клеток моноцитарного происхождения (клеток Лангерганса), характеризующихся наличием гранул Бирбека*** по результатам электронной микроскопии.
P.S. *** Клетки Лангерганса содержат развитые органеллы и особые мембранные гранулы Бирбека в форме теннисной ракетки, содержащие лангерин.
Наличие двухслойных гранул Бирбека в форме теннисной ракетки в цитоплазме патологических клеток является диагностическим признаком гистиоцитоза из клеток Лангерганса. Гранула Бирбека экспрессирует недавно АГн лангергин (CD207), который участвует в презентации АГн Т-лимфоцитам. Было установлено, что экспрессия CD207 наблюдается при гистиоцитозе из клеток Лангерганса постоянно и, т.о., является еще одним надежным диагностическим маркером.
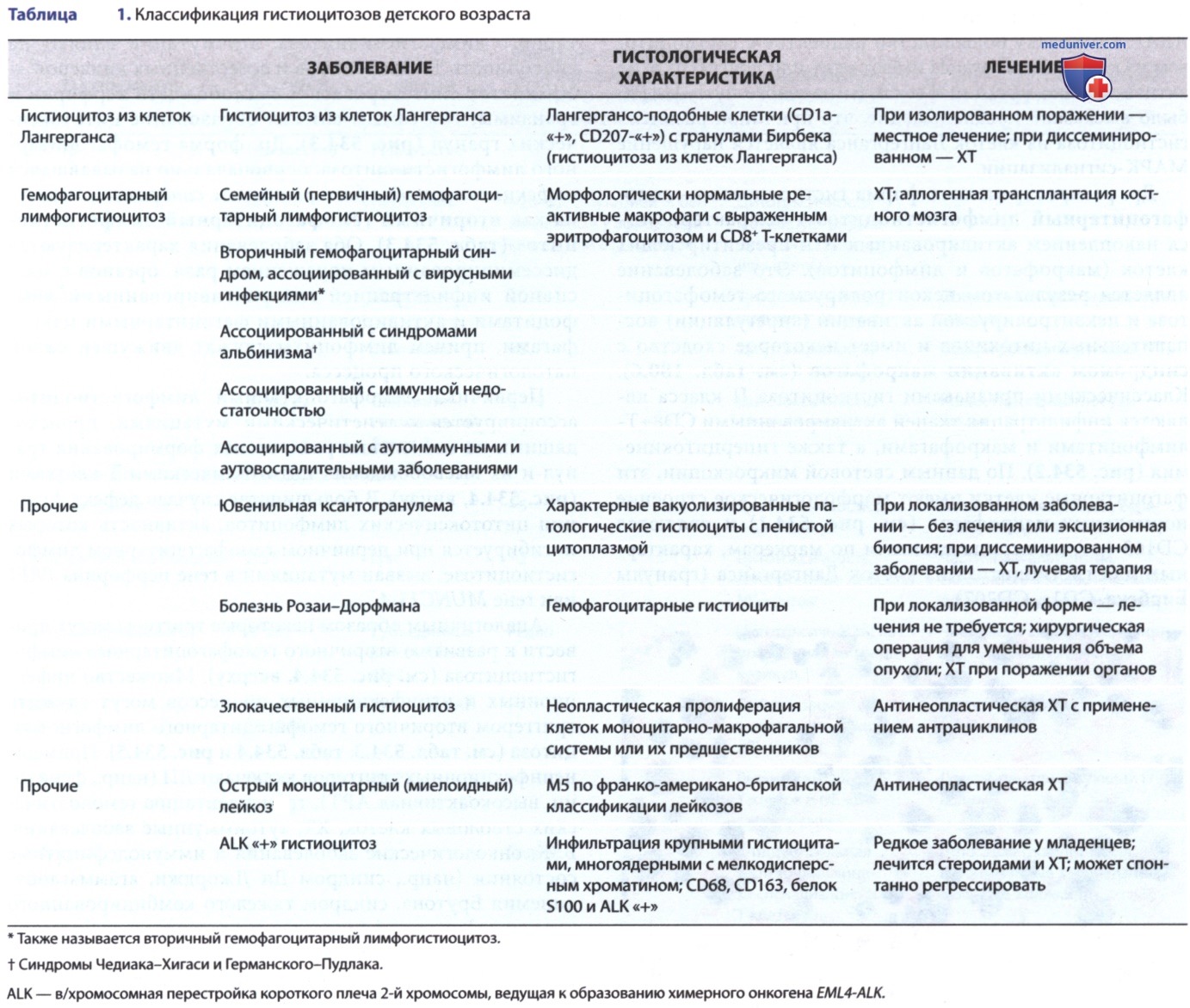
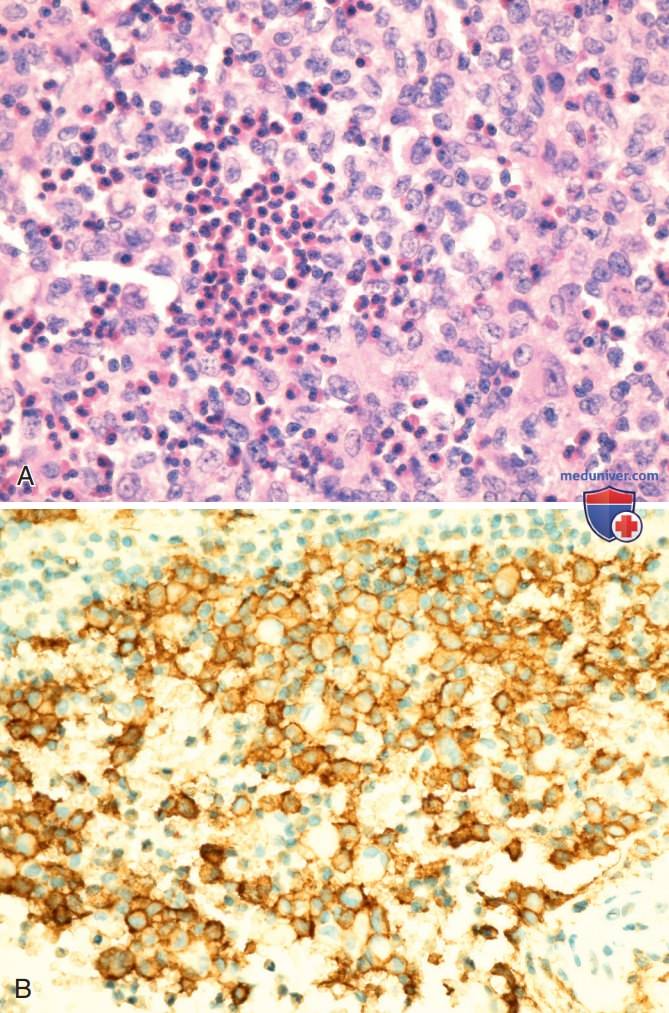
Более вероятно, что патологические клетки на самом деле не являются дифференцированными клетками Лангерганса, а скорее незрелыми миелоидными клетками, которые, возможно, перестали развиваться. Окончательный диагноз «гистиоцитоз из клеток Лангерганса» устанавливается на основании «+» результатов гистопатологического исследования на экспрессию CD1а патологическими клетками (рис. 1). Патологические клетки необходимо отличать от нормальных эпидермальных клеток Лангерганса, которые также являются CD1a-«+», но расположены далеко друг от друга и не являются диагностическим признаком гистиоцитоза из клеток Лангерганса.
Рисунок 1. Гистиоцитоз из клеток Лангерганса: А — гистопатология гистиоцитоза из клеток Лангерганса. Изображение эозинофильной гранулемы с литическими очагами в головке бедренной кости. Смешанный инфильтрат со множественными клетками, гистиоцитоза из клеток Лангерганса с характерными бороздчатыми ядрами и многочисленными эозинофилами; В — окрашивание клеток на CD1a, диагностический маркер гистиоцитоза из клеток Лангерганса
Соотношение гранул Бирбека, содержащих CD1a-«+» клетки, а также лимфоцитов, гранулоцитов, моноцитов и эозинофилов в периферических пораженных тканях (напр., в коже, ЛУ, костях) м.б. различным.
В некоторых случаях гистиоцитоза из клеток Лангерганса наблюдается клональный характер пролиферации клеток в отдельных очагах поражения. Важно отметить, что у многих пациентов с гистиоцитозом из клеток Лангерганса была выявлена активирующая соматическая мутация в гене BRAF**** (V600E).
P.S. **** Мутация BRAF V600E представляет собой наиболее частую аберрацию, обнаруживаемую при папиллярном раке ЩЖ (40-50% случаев). Также данная мутация встречается в 20-40% случаев низкодифференцированных карцином и 30-40% случаев анапластических карцином.
Исследования пациентов с отрицательным результатом на BRAFV600E выявили мутации в др. генах МАРК*, включая МАР2К1 и ARAF. Поскольку большинство пациентов с гистиоцитозом из клеток Лангерганса имеют одну или другую из этих активирующих мутаций в генах сигнального пути МАРК, было высказано предположение, что причиной развития гистиоцитоза из клеток Лангерганса является нарушение МАРК-сигнализации.
P.S. * МАРК mitogen-activated protein kinase — митоген-активируемая протеинкиназа — группа мультифункциональных в/клеточных сигнальных путей, содержащих одну из митоген-активируемых протеинкиназ и контролирующих транскрипцию генов, метаболизм, пролиферацию и подвижность клеток, апоптоз и др. процессы.
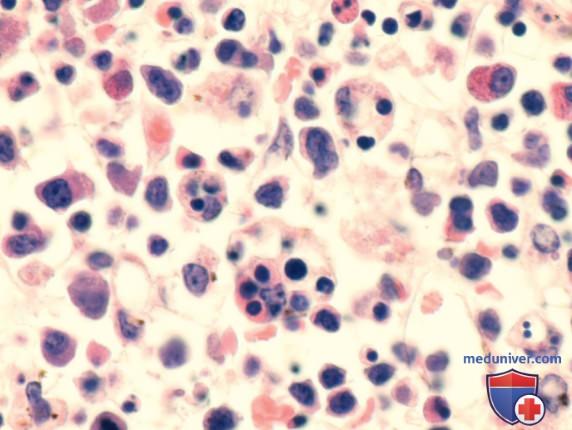
Др. распространенная форма гистиоцитоза — гемофагоцитарный лимфогистиоцитоз — характеризуется накоплением активированных АГн-презентирующих клеток (макрофагов и лимфоцитов). Это заболевание является результатом неконтролируемого гемофагоцитоза и неконтролируемой активации (апрегуляции) воспалительных цитокинов и имеет некоторое сходство с синдромом активации макрофагов. Классическими признаками гистиоцитоза II класса являются инфильтрация тканей активированными CD8+T-лимфоцитами и макрофагами, а также гиперцитокинемия (рис. 2).
Рисунок 2. Аспират костного мозга ребенка с семейным (генетически подтвержденным) гемофагоцитарным лимфогистиоцитозом. Видны многочисленные характерные гемофагоцитарные клетки (которые являются CD163-«+» макрофагами), поглощающие разл. элементы крови
По данным световой микроскопии, эти фагоцитарные клетки имеют морфологическое строение нормальных макрофагов (см. рис. 1) и являются CD163-«+», но отрицательными по маркерам, характерным для гистиоцитоза из клеток Лангерганса (гранулы Бирбека, CD1a, CD207).
Клинические картины двух основных форм гемофагоцитарного лимфогистиоцитоза неотличимы друг от друга, но в каждом случае необходимо провести ДД, т.к. от точного диагноза зависят тактика лечения и прогноз. Первичный гемофагоцитарный лимфогистиоцитоз, ранее называемый семейным эритрофагоцитарным лимфогистиоцитозом, сейчас известен как семейный гемофагоцитарный лимфогистиоцитоз. Это заболевание представляет собой АуР-нарушение и составляет 25% от всех случаев гемофагоцитарного лимфогистиоцитоза (табл. 2).
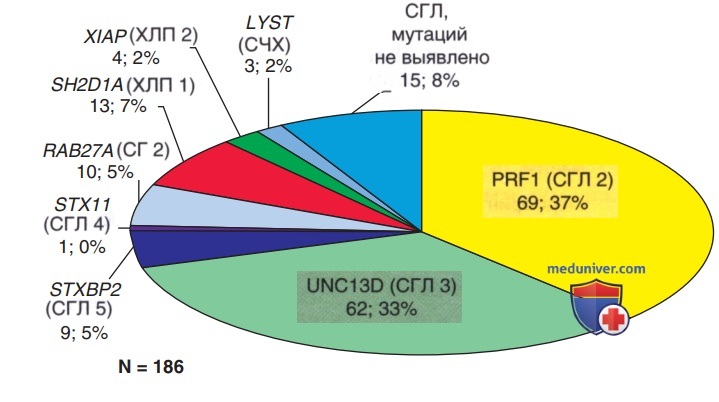
Были выявлены гены для 4 из 5 семейных синдромов гемофагоцитарного лимфогистиоцитоза и др. наследственных форм гемофагоцитарного лимфогистиоцитоза; эти мутации влияют на способность Т-лимфоцитов и естественных киллеров — NK-клеток синтезировать и высвобождать перфорин и гранзимы, тем самым снижая образование цитотоксических гранул (рис. 3). Др. форма гемофагоцитарного лимфогистиоцитоза, первоначально называвшаяся инфекционным гемофагоцитарным синдромом, известна как вторичный гемофагоцитарный лимфогистиоцитоз (табл. 3). Оба заболевания характеризуются диссеминированным поражением разл. органов с массивной инфильтрацией гиперактивированными лимфоцитами и активированными фагоцитарными макрофагами, причем лимфоциты служат движущей силой патологического процесса.
Рисунок 3. Распределение генетических подтипов в группе из 171 пациента с семейным гемофагоцитарным лимфогистиоцитозом или заболеванием, связанным с семейным гемофагоцитарным лимфогистиоцитозом. Для каждого подтипа указано название гена, аббревиатура подтипа заболевания, количество случаев в группе и процент от общего числа. Кроме того, в группу с семейным гемофагоцитарным лимфогистиоцитозом относится подгруппа из 15 пациентов с семейным рецидивом или рефрактерным/рецидивирующим заболеванием после специфической терапии, а также пациенты с неоднократно документированными тяжелыми функциональными дефектами по результатам исследования на дегрануляцию или цитотоксичность. СГЛ — семейный гемофагоцитарный лимфогистиоцитоз; СЧХ — синдром Чедиака-Хигаси — редкое иммунодефицитное состояние с АуР-типом наследования, обусловленное мутацией в гене LYST/CHS1; ХЛП — Х-сцепленный лимфопролиферативный синдром — это комбинированный первичный иммунодефицит, характеризующийся атипичной реакцией на инфекцию ВЭБ, вследствие чего развивается гемофагоцитоз, дисгаммаглобулинемией, аутоиммунной патологией и злокачественной лимфопролиферацией. XIAP — X-linked inhibitor of apoptosis
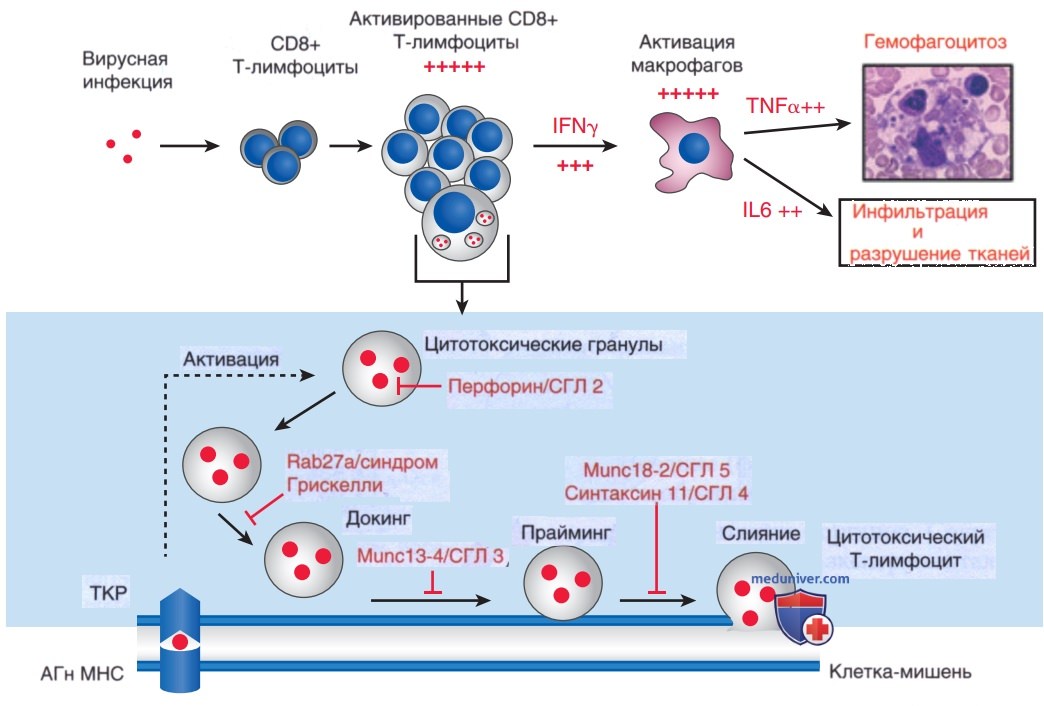
Первичный гемофагоцитарный лимфогистиоцитоз ассоциируется с генетическими мутациями, происходящими на нескольких разл. этапах формирования гранул и их высвобождения цитотоксическими Т-клетками (рис. 4, внизу). В большинстве случаев дефект функции цитотоксических лимфоцитов, активность которых ингибируется при первичном гемофагоцитарном лимфогистиоцитозе, вызван мутациями в гене перфорина PRF1 или гене MUNC13-4.
Рисунок 4. Врожденные дефекты цитотоксической активности лимфоцитов. Вверху: схема иммунных механизмов, приводящих к развитию гемофагоцитарного синдрома. После вирусной инфекции антиген (АГн)-специфические CD8+ Т-лимфоциты подвергаются массивной экспансии и активации и секретируют высокий уровень интерферона-γ (IFN-y). Огромное число активированных эффекторных клеток вызывает чрезмерную активацию макрофагов и выработку провоспалительных цитокинов, включая фактор некроза опухоли-a (ФНО-α) и интерлейкин-6 (IL-6). Элементы крови (тромбоциты, эритроциты и полиморфноядерные клетки, изображенные на рисунке) спонтанно фагоцитируются макрофагами. Активированные лимфоциты и макрофаги проникают в разл. органы, что приводит к массивному некрозу тканей и отказу органов. Внизу: генетические дефекты, вызывающие гемофагоцитарный лимфогистиоцитарный синдром, влияют на определенный этап цитотоксического механизма: содержание гранул, докинг, прайминг или слияние. Показаны только дефекты, вызывающие синдром Грисцелли и семейный гемофагоцитарный лимфогистиоцитоз
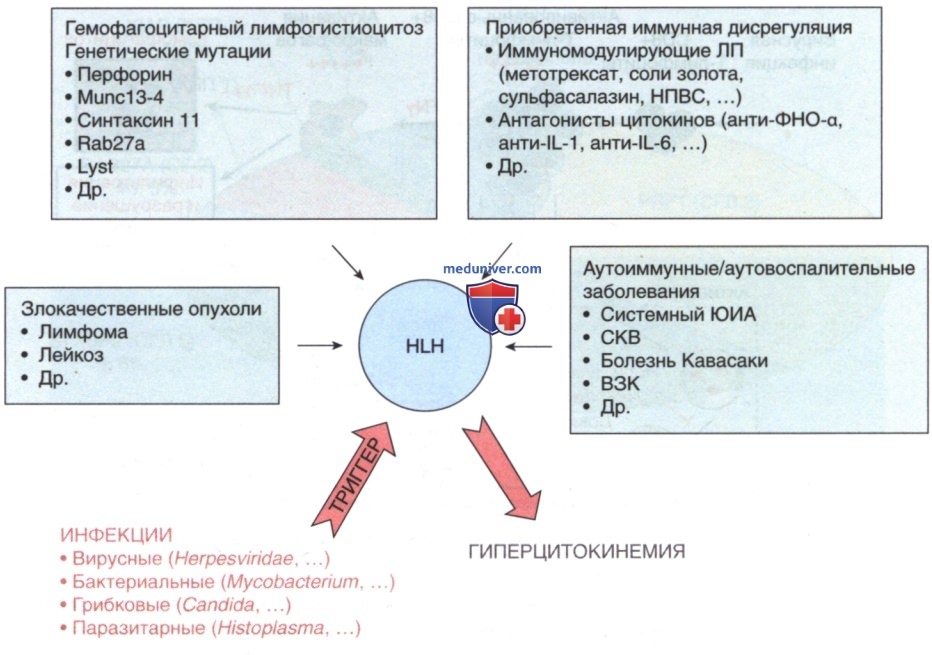
Аналогичным образом некоторые триггеры могут привести к развитию вторичного гемофагоцитарного лимфогистиоцитоза (см. рис. 4, вверху). Множество инфекционных и неинфекционных процессов могут служить триггером вторичного гемофагоцитарного лимфогистиоцитоза (см. табл. 3; табл. 4 и рис. 5). Примеры неинфекционных триггеров включают ЛП (напр., фенитоин, высокоактивная APT), трансплантацию гемопоэтических стволовых клеток, XT, аутоиммунные заболевания, ВЗК, онкологические заболевания и иммунодефицитные состояния (напр., синдром Ди Джорджи, агаммаглобулинемия Брутона, синдром тяжелого комбинированного иммунодефицита, хроническая гранулематозная болезнь).
Рисунок 5. Гемофагоцитарный лимфогистиоцитоз представляет собой группу разл. заболеваний, которые характеризуются тяжелой гиперцитокинемией и иммунопатологическими состояниями, угрожающими жизни. Гемофагоцитарный лимфогистиоцитоз м.б. вызван мутациями в генах, отвечающих за гранулоопосредованную цитотоксичность, но также м.б. приобретенным на фоне множества разных основных аутоиммунных/ аутовоспалительных заболеваний или злокачественных новообразований, с возможным облегчением после иммуномодулирующей терапии. Клинические проявления гемофагоцитарного лимфогистиоцитоза обычно вызываются инфекцией
Помимо двух вышеописанных наиболее распространенных форм детского гистиоцитоза (гистиоцитоза из клеток Лангерганса и гемофагоцитарного лимфогистиоцитоза), в эту же группу входят и др., более редкие заболевания. Ювенильная ксантогранулема характеризуется наличием в очагах поражения вакуолизированных гистиоцитов с пенистой цитоплазмой, которые развиваются в смешанные гранулемы, содержащие также эозинофилы, лимфоциты и др. клетки. Болезнь Эрдгейма-Честера преимущественно поражает взрослых. Поверхностные маркеры указывают на связь между гистиоцитозом из клеток Лангерганса, ювенильной ксантогранулемой и болезнью Эрдгейма-Честера; все 3 заболевания развиваются из дендритных клеток и ассоциируются с мутациями BRAFV600E в пораженных клетках.
Др. редкой формой гистиоцитоза является болезнь Розаи-Дорфмана, также известная как синусовый гистиоцитоз с массивной лимфаденопатией.
Болезнь Розаи-Дорфмана характеризуется аккумуляцией гемофагоцитарных гистиоцитов в синусах ЛУ, хотя может развиваться и экстранодальное поражение. Наконец, к третьему классу гистиоцитоза детского возраста относится группа ЗНО из клеток моноцитарномакрофагальной линии. Согласно этому определению, острый моноцитарный лейкоз и истинный злокачественный гистиоцитоз относятся к гистиоцитозам класса III. Истинные новообразования из клеток Лангерганса встречаются крайне редко.
б) Гистиоцитоз из клеток Лангерганса:
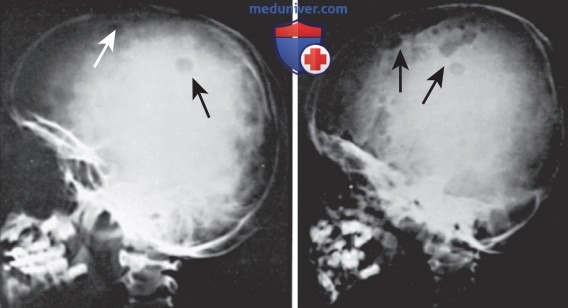
1. Клинические проявления. Клиническая картина гистиоцитоза из клеток Лангерганса чрезвычайно разнообразна. У 80% пациентов наблюдаются поражения костей, а у детей >5 лет гистиоцитоз из клеток Лангерганса может локализоваться только в костях. Поражения костей м.б. одиночными или множественными, но чаще всего поражается череп (рис. 6). В некоторых случаях также могут поражаться таз, бедренная кость, позвонки, верхняя и нижняя челюсти. Гистиоцитоз из клеток Лангерганса может протекать бессимптомно или сопровождаться болью и локальным отеком. Поражение позвонков может привести к коллапсу тела позвонка, который виден на рентгенограмме.
Рисунок 6. Рентгенограммы черепа пациентов с гистиоцитозом из клеток Лангерганса. Слева: рентгенограмма черепа ребенка >2 лет. Стрелками указано изолированное поражение кости. Исход достаточно благоприятный. Справа: рентгенограмма черепа ребенка <2 лет. Стрелками указано обширное поражение кости. Кроме того, у пациента наблюдались лихорадка, анемия, обширное поражение кожи, генерализованная лимфоаденопатия, гепатоспленомегалия и инфильтраты в легких. Несмотря на противоопухолевую химиотерапию, исход был летальным. Данные случаи представляют собой крайние варианты гистиоцитоза из клеток Лангерганса
Коллапс тела позвонка, в свою очередь, может привести к вторичной компрессии спинного мозга. В плоских и трубчатых костях возникают остеолитические поражения с четкими границами без признаков регенерации новой костной ткани. При поражении опорных длинных трубчатых костей могут возникать патологические переломы. Поражение сосцевидного отростка сопровождается заболеванием среднего уха с выходом гнойного отделяемого. Из-за разрушения костной ткани в нижней и верхней челюсти на рентгенограмме может казаться, что некоторые зубы не зафиксированы в кости. При выборе подходящей терапии возможно полное излечение.
Примерно у 50% пациентов во время болезни развивается поражение кожи (изолированное или как часть мультисистемного поражения), обычно в виде трудно поддающегося лечению чешуйчатого, папулезного, себорейного дерматита волосистой части головы, паховой области, подмышек или области позади ушных раковин (рис. 7 и 8). Поражения могут распространяться на спину, ладони и ступни. Высыпания на коже имеют петехиальный или геморрагический характер, даже при отсутствии тромбоцитопении. У 33% пациентов наблюдается локализованная или диссеминированная лимфоаденопатия, а у 20% — гепатоспленомегалия. Могут наблюдаться поражения печени разл. степени тяжести, включая желтуху и асцит.
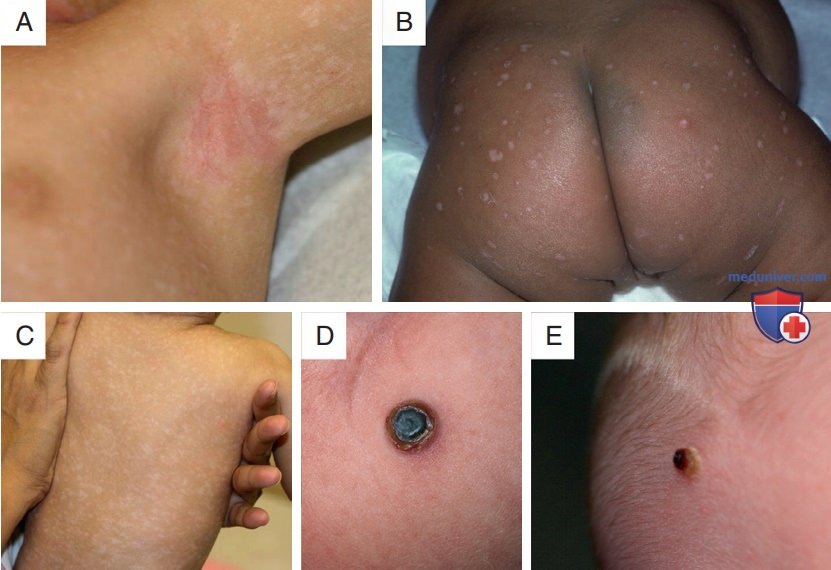
Рисунок 7. Разнообразные проявления гистиоцитоза из клеток Лангерганса на коже: А — экзематозный дерматит; В — гипопигментированные эрозированные папулы; C — гипомеланозные макулы; D и E — папулы и нодулы, покрытые коркой. По внешнему виду нельзя сказать о наличии или отсутствии мультисистемного заболевания. Несмотря на схожий внешний вид, у пациента с изображения г было изолированное поражение, в то время как у пациента с изображения д наблюдалось поражение органов
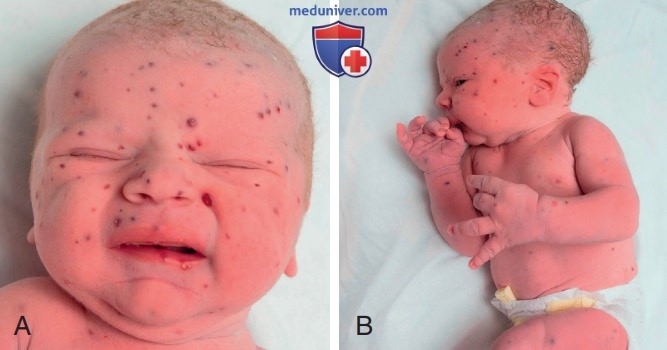
Рисунок 8. Гистиоцитоз из клеток Лангерганса, проявляющийся в виде сыпи на коже по типу «черничного кекса» у новорожденного. Множественные твердые, не бледные, пурпурные папулы, поражающие голову и шею (А), а также тело (В)
В некоторых случаях развивается экзофтальм, который часто бывает двусторонним и обусловлен гранулематозным воспалением ретроорбитальной ткани. Инфильтративное поражение слизистых оболочек десен напоминает кандидоз. У 30-40% пациентов развивается отит среднего уха, который иногда может привести к глухоте. У 10-15% пациентов при РОГК обнаруживаются инфильтраты в легких. Поражения легких могут варьироваться от диффузного фиброза и диссеминированных узелковых инфильтратов до диффузных кистозных изменений (рис. 9). В редких случаях развивается пневмоторакс. При сильном поражении легких может возникнуть тахипноэ и прогрессирующая ДН.
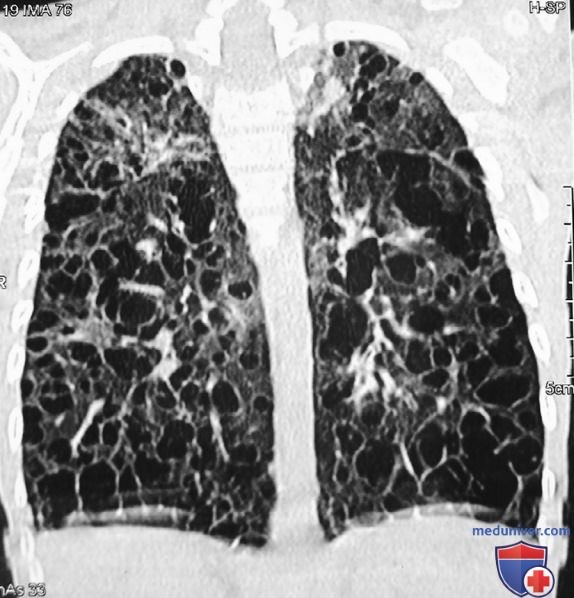
Рисунок 9. Диффузные кисты с деструкцией легочной паренхимы в обоих легочных полях на компьютерно-томографическом изображении с высоким разрешением (легочный режим) в корональной проекции
Дисфункция гипофиза или поражение гипоталамуса сопровождаются задержкой роста. Кроме того, у пациентов м.б. несахарный диабет, поэтому перед проведением биопсии необходимо убедиться, что пациенты способны концентрировать мочу. В редких случаях может возникнуть пангипопитуитаризм, а также первичный гипотиреоз вследствие инфильтрации ЩЖ.
Системные проявления тяжелой формы заболевания включают лихорадку, МТ, общее недомогание, раздражительность и задержку развития. Такие системные проявления позволяют отличить пациентов из группы высокого риска летального исхода (т.е. с поражением органов риска) от пациентов из группы низкого риска летального исхода (т.е. без системных проявлений; пациенты без поражения «органов риска»). «Органами риска» являются печень, селезенка и кроветворная система (костный мозг). Легкие не считаются органом риска.
Определение степени поражения органов риска важно для принятия решения об интенсивности лечения и учитывается в стандартных подходах к лечению гистиоцитоза из клеток Лангерганса в протоколах Гистиоцитарного общества. Поражение костного мозга может вызывать анемию и тромбоцитопению.
К двум редким, но серьезным проявлениям гистиоцитоза из клеток Лангерганса относятся поражение печени (которое может привести к фиброзу и циррозу) и ЦНС, характеризующееся атаксией, дизартрией и др. неврологическими симптомами. Поражение печени связано с мультисистемным заболеванием, которое во многих случаях уже проявляется на момент постановки диагноза гистиоцитоза из клеток Лангерганса. Прогрессирующее поражение ЦНС в виде глиоза, напротив, может проявляться только через много лет после постановки основного диагноза. Лечение для данного поражения ЦНС на сегодняшний день не установлено.
Ни одно из описанных выше проявлений не ассоциируется с клетками Лангерганса, гранулами Бирбека, CD1a-позитивностью или любыми др. признаками инфильтрации клетками Лангерганса, что вызывает вопросы о патогенезе этих состояний.
После биопсии, самого простого диагностического метода при поражении кожи или костей, необходимо провести тщательное клиническое и лабораторное обследование каждого пациента, которое должно включать следующие исследования: ОАК, функциональные пробы печени, исследование свертываемости крови, исследование скелета, РОГК и измерение осмоляльности мочи. Если по результатам лабораторных исследований возникает подозрение о поражении какой-либо системы органов, перед началом лечения необходимо провести тщательное обследование этой системы.
2. Лечение и прогноз. Клиническое течение заболевания с поражением одного органа (обычно костей, ЛУ или кожи) обычно доброкачественное, с высокой вероятностью спонтанной ремиссии. Поэтому лечение должно быть минимальным и направленным на ограничение роста новообразования, которое может привести к необратимому повреждению органа, прежде чем оно спонтанно разрешится. Для этой цели, напр., в случае поражения кости, показан кюретаж или, реже, введение кортикостероидов или местная низкодозированная лучевая терапия (5-6 Гр).
Мультисистемное заболевание необходимо лечить с помощью системной мультиагентной XT по нескольким разл. схемам на основе винбластина и ГКС. Схемы на основе этих ЛП оказались очень эффективными в лечении гистиоцитоза из клеток Лангерганса. Этопозид был исключен из стандартного лечения мультисистемной формы гистиоцитоза из клеток Лангерганса, которая должна лечиться несколькими агентами для того, чтобы снизить вероятность летального исхода, частоту рецидивов и долгосрочных эффектов терапии. Частота ответа на терапию довольно высока, а смертность при тяжелой форме гистиоцитоза из клеток Лангерганса была существенно снижена благодаря мультиагентной ХТ, особенно в случае точного и раннего диагноза. Согласно последним данным, выживаемость при тяжелом мультисистемном заболевании (с поражением органов риска) после длительной непрерывной терапии составила >85%, а частота развития рецидивов снизилась.
Экспериментальная терапия показана только при неэффективности стандартных схем лечения, напр., очень маленьким детям с мультисистемным заболеванием и дисфункцией органов, которые не отвечают на мультиагентную начальную терапию, а также с реактивацией поражения любых органов риска, за исключением легких. Современные подходы к лечению включают иммуносупрессивную терапию циклоспорином/иммуноглобулином антитимоцитарным и, возможно, иматинибом, 2-хлрро-дезоксиаденозином, клофарабином, а также трансплантацию стволовых клеток. С открытием мутации BRAF V600E, вызывающей гиперактивацию сигнального пути МАРК в гистиоцитарных клетках, фармакологическое ингибирование BRAF и МЕК изучаются в качестве терапевтических подходов для лечения резистентной формы заболевания.
Поздние (фиброзные) осложнения, в печени или легких, необратимы и для окончательного излечения требуют трансплантации органов. Современные подходы к лечению и протоколы экспериментальных исследований методов лечения гистиоцитоза из клеток Лангерганса и гемофагоцитарного лимфогистиоцитоза можно найти на сайте Гистиоцитарного Общества. Нерешенной проблемой остается лечение (обычно позднее) прогрессирующего и трудноизлечимого нейроде-генеративного синдрома, ассоциированного с гистиоцитозом из клеток Лангерганса.
в) Гемофагоцитарный лимфогистиоцитоз. См. раздел «Гистопатологическая классификация» в начале данной статьи на сайте.
1. Клинические проявления. Семейный гемофагоцитарный лимфогистиоцитоз и вторичный гемофагоцитарный лимфогистиоцитоз имеют сходные клинические проявления, включающие генерализованный патологический процесс, чаще всего с лихорадкой (90-100%), макулопапулезной и/или петехиальной сыпью (10-60%), ↓ МТ и раздражительностью. Начальная клиническая картина может различаться, но почти всегда эти заболевания проявляются очень тяжелыми симптомами, а в случае вторичного гемофагоцитарного лимфогистиоцитоза клиническая картина м.б. замаскирована основным заболеванием. Острое начало заболевания включает септический шок, ОРДС, судороги и кому (из-за инфильтрации ЦНС). Др. часто встречающиеся признаки обусловлены поражением костного мозга и панцитопенией или дисфункцией печени.
Хотя обе формы гистиоцитоза встречаются в любом возрасте, первичный гемофагоцитарный лимфогистиоцитоз обычно развивается у детей до 1-2 лет, а вторичный гемофагоцитарный лимфогистиоцитоз чаще всего проявляется в более старшем возрасте. Физикальное обследование часто выявляет гепатоспленомегалию (70-100%), лимфаденопатию (20-50%), ОРДС (40-90%), желтуху и симптомы поражения ЦНС (50%), сходные с симптомами асептического менингита или острого демиелинизирующего энцефаломиелита. На MPT-изображениях в режимах Т2 и FLAIR могут визуализироваться гиперинтенсивные очаги в сером и белом в-ве ГМ, а также в супратенториальных и инфратенториальных областях.
Плеоцитоз в СМЖ (50-90%), ассоциированный с поражением ЦНС при первичном гемофагоцитарном лимфогистиоцитозе, характеризуется такими же фагоцитирующими макрофагами, которые обнаруживаются в периферической крови или костном мозге. Первичный гемофагоцитарный лимфогистиоцитоз также обычно ассоциируется с тяжелым иммунодефицитом.
Диагностика гемофагоцитарного лимфогистиоцитоза происходит в 2 этапа. На первом этапе оцениваются 8 клинических и лабораторных признаков, причем наличие 5 из 8 признаков является ДК для гемофагоцитарного лимфогистиоцитоза. Эти 8 признаков, сформулированные Обществом по изучению гистиоцитоза, включают лихорадку, спленомегалию, цитопению 2 клеточных линий (в 90-100% случаев), гипертриглицеридемию (80-100%) или гипофибриногенемию (65-85%), гиперферритинемию (>500, но часто >10 000), чрезвычайно высокое содержание растворимого CD25 (рецептор IL-2), сниженную или нулевую активность NK-клеток, а также признаки гемофагоцитоза в костном мозге, СМЖ или ЛУ (табл. 5). На втором этапе проводится генетический анализ на наличие специфических мутаций.
Несмотря на то, что обычно на проведение этого анализа требуется некоторое время, не стоит откладывать начало лечения (рис. 10). По результатам генетического анализа и семейному анамнезу определяют, является ли заболевание первичной (АуР) или вторичной формой гемофагоцитарного лимфогистиоцитоза.
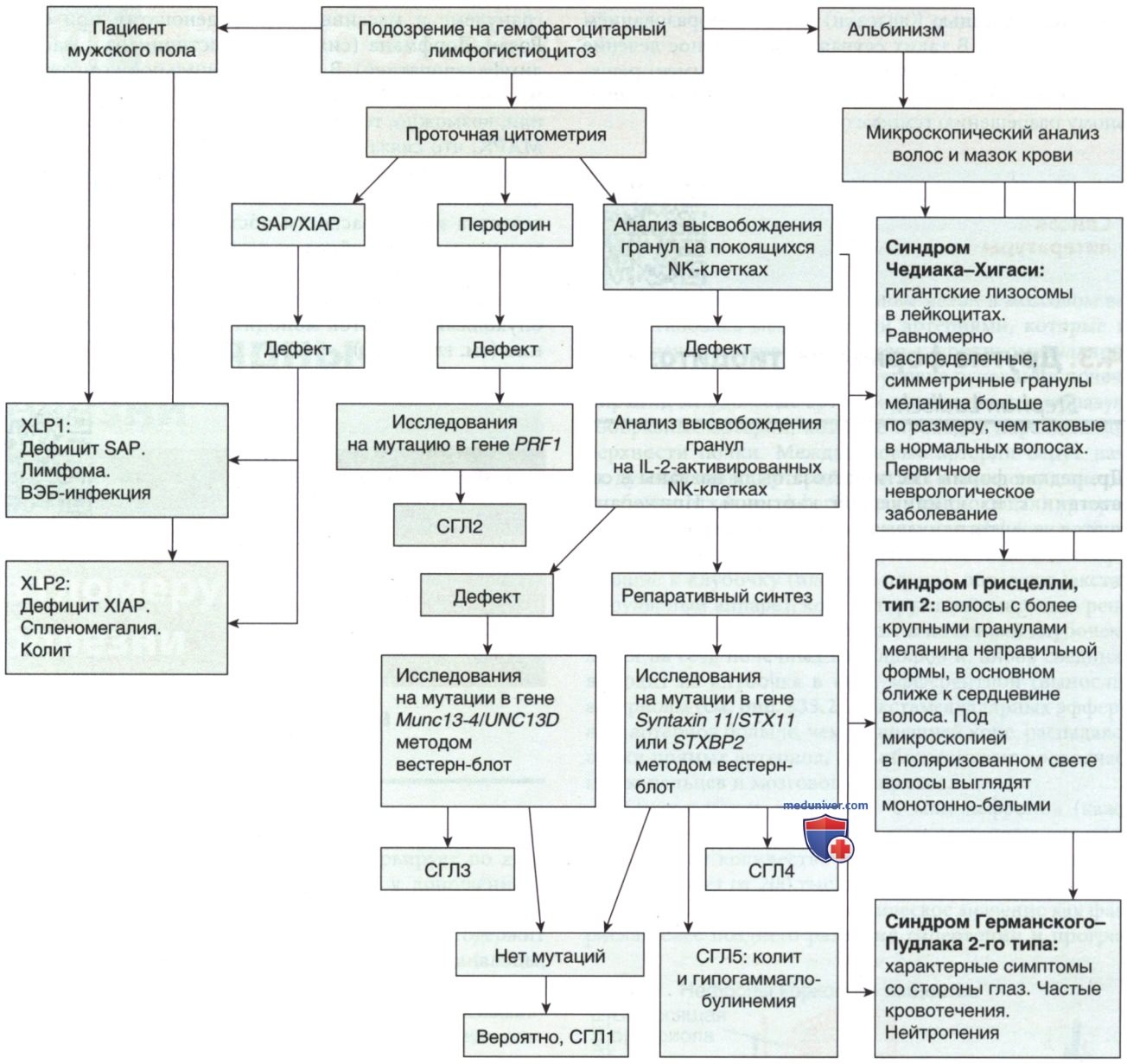
Рисунок 10. Алгоритм выявления генетических причин развития гемофагоцитарного лимфогистиоцитоза. Данный алгоритм основан на данных проточной цитометрии: все пациенты с подозрением на гемофагоцитарный лимфогистиоцитоз, независимо от возраста и клинической картины, должны быть обследованы на экспрессию перфорина и высвобождение гранул. Все пациенты мужского пола должны быть обследованы на экспрессию белка, ассоциированного с сигнальной молекулой активации лимфоцитов, а также белка Х-сцепленного ингибитора апоптоза. Для пациентов с клиническими проявлениями альбинизма необходимо провести микроскопический анализ волос и мазка крови для дифференциальной диагностики синдрома Чедиака-Хигаси, синдрома Грисцелли и синдрома Германского-Пудлака. На основании выявленного дефекта экспрессии определенного белка для подтверждения диагноза необходимо провести молекулярную оценку соответствующего гена. СГЛ: семейный гемофагоцитарный лимфогистиоцитоз; NK (Natural killer) — естественные киллеры; SAP (SLAM-Associated Protein) — сигнальная молекула активации лимфоцита; XIAP (X-linked inhibitor of apoptosis) — белок Х-сцепленный ингибитор апоптоза; XLP (X-linked lymphoproliferative syndrome) — Х-сцепленный лимфопролиферативный синдром
Гемофагоцитоз не является специфическим проявлением гемофагоцитарного лимфогистиоцитоза и должен рассматриваться в контексте ДК. Клинические или лабораторные различия между первичной и вторичной формами гемофагоцитарного лимфогистиоцитоза отсутствуют.
В некоторых подгруппах гемофагоцитарного лимфогистиоцитоза результат анализа на перфорин м.б. в норме. Аналогичным образом, в некоторых случаях первичного семейного гемофагоцитарного лимфогистиоцитоза не определяется ни одной из известных генных мутаций, ассоциированных с данным заболеванием.
При отсутствии (1) документально подтвержденного генетического дефекта в сочетании с дефектной цитотоксичностью NK-клеток или (2) выраженного гемофагоцитоза следует с осторожностью ставить диагноз вторичного гемофагоцитарного лимфогистиоцитоза, учитывая необходимость применения цитотоксической XT. Неспецифические признаки (указывающие на воспаление), используемые в качестве критериев при диагностике гемофагоцитарного лимфогистиоцитоза, могут также наблюдаться при заболеваниях, не всегда связанных с гемофагоцитозом (напр., при массивной ОРВИ с соответствующей активацией Т-клеток), при которых цитотоксическая и иммуносупрессивная терапии, применяемые для лечения гемофагоцитарного лимфогистиоцитоза, м.б. противопоказаны.
Синдром активации макрофагов, особенно в сочетании с системным ЮИА или инфекцией, имеет много общих характеристик с гемофагоцитарным лимфогистиоцитозом. Действительно, полноэкзомное секвенирование пациентов с системным ЮИА или пациентов с летальным исходом от гриппа выявило более высокую, чем ожидалось, частоту встречаемости генов гемофагоцитарного лимфогистиоцитоза. В ДД гемофагоцитарного лимфогистиоцитоза входят также сепсис, болезнь Вольмана, остеопетроз, аутоиммунный лимфопролиферативный синдром, неонатальный гемохроматоз, болезнь Гоше, комбинированный иммунодефицит и общий вариабельный иммунодефицит.
2. Лечение и прогноз. Лечение первичного гемофагоцитарного лимфогистиоцитоза (АуР-генетическое заболевание или семейное происхождение) включает комбинированную терапию ЛП этопозида, ГКС, циклоспорина и интратекального метотрексата, согласно описанию в современных протоколах HLH-1994 и HLH-2004 Гистиоцитарного Общества. Следует подчеркнуть, что панцитопения и наличие инфекции не являются противопоказаниями к цитотоксической терапии. Некоторые исследователи рекомендуют антитимоцитарный глобулин и циклоспорин в качестве поддерживающей терапии. Цель такой терапии — дождаться момента, когда можно будет провести трансплантацию стволовых клеток, которая на сегодняшний день является единственным известным методом лечения первичного гемофагоцитарного лимфогистиоцитоза, при котором частота излечения составляет >60%.
С помощью одной только XT нельзя достичь длительного излечения первичного гемофагоцитарного лимфогистиоцитоза, т.к. без трансплантации стволовых клеток в конечном итоге наступает летальный исход.
При вторичном гемофагоцитарном лимфогистиоцитозе необходимо выявить и провести успешное лечение основного заболевания (напр., инфекции или ЗНО). Диагностическое различие между первичной и вторичной формами гемофагоцитарного лимфогистиоцитоза иногда м.б. основано на остром начале вторичного гемофагоцитарного лимфогистиоцитоза при наличии лабораторно подтвержденной инфекции. В этом случае лечение основной инфекции сочетается с сопроводительной терапией. Если диагноз поставлен на фоне ятрогенного иммунодефицита, следует отменить иммуносупрессивное лечение и начать поддерживающую терапию наряду со специфической терапией основной инфекции.
Многие пациенты имеют очень хороший прогноз и без дополнительного специфического лечения, кроме лечения инфекции, вызвавшей заболевание. Однако, если не удается документально подтвердить наличие инфекции или др. причины, поддающейся лечению, и если клиническая картина тяжелая, прогноз при вторичном гемофагоцитарном лимфогистиоцитозе такой же плохой, как и при первичном. Такие пациенты должны получать первоначальную XT в течение 8 нед, включая этопозид, даже при наличии цитопении. Как при первичном, так и при вторичном гемофагоцитарном лимфогистиоцитозе цитотоксическое действие этопозида на макрофаги прерывает выработку цитокинов, гемофагоцитоз и накопление макрофагов, что обусловливает патогенез инфекционно-ассоциированного гемофагоцитарного синдрома.
Вторичный гемофагоцитарный лимфогистиоцитоз м.б. обусловлен широким спектром инфекционных агентов, включая вирусы (напр., ЦМВ, ВЭБ, ВГЧ 6), грибы, простейшие и бактерии, которые м.б. триггерами гемофагоцитарного лимфогистиоцитоза, часто на фоне иммунодефицита (см. табл. 3). Пациентов с иммунодефицитом и гемофагоцитозом необходимо тщательно обследовать на наличие инфекции. Тот же синдром м.б. выявлен в сочетании с ревматическим заболеванием (напр., СКВ, болезнью Кавасаки) или новообразованием (напр., лейкозом). В таких случаях эффективное лечение основного заболевания (инфекции или ЗНО) имеет решающее значение и может само по себе привести к окончательному разрешению гемофагоцитоза.
г) Другие формы гистиоцитоза. Др. редкие формы гистиоцитоза были названы в соответствии с их клинической картиной. Примерами являются ксантогранулема при ювенильной ксантогранулеме и массивная лимфаденопатия при болезни Розаи-Дорфмана (синусовый гистиоцитоз с массивной лимфаденопатией). В случае ювенильной ксантогранулемы может потребоваться системная цитотоксическая XT или, возможно, терапия ингибиторами сигнального пути МАРК, что связано с наличием BRAF-мутации. Болезнь Розаи-Дорфмана обычно не требует лечения, хотя массивная лимфаденопатия может потребовать вмешательства из-за опасности обструкции органов.
Острый моноцитарный лейкоз и истинный злокачественный гистиоцитоз относятся к гистиоцитозам III класса, поскольку они однозначно являются злокачественными опухолями из клеток моноцитарно-макрофагальной линии.