а) Кожные ямки. Вдавления над костными выступами и в крестцовой области, иногда в сочетании с ямками и складками, могут возникать у здоровых детей и быть проявлениями дисморфологических синдромов. Ямки на коже формируются в период в/утробного развития в результате нарушения развития ПЖК в области, сдавленной между острым костным выступом и стенкой матки.
Кожные ямки также могут присутствовать над областью гипоплазии кости. Двусторонние ямки над акромиальным отростком — изолированный симптом, но также могут наблюдаться при делеции длинного плеча хромосомы 18. Кожные ямки чаще обнаруживаются над надколенником при врожденной краснухе, над боковыми сторонами коленных и локтевых суставов при аплазии мышц брюшной стенки, I над передней поверхностью большеберцовой кости при кампомелической карликовости и в форме буквы Н на подбородке при синдроме «свистящего человека».
Крестцовые ямки встречаются часто и обычно не сопровождаются др. симптомами. Также их можно увидеть при многочисленных синдромах или в сочетании со скрытым незаращением дужек позвонков и диастомиелией. Обнаружение сопутствующего объемного образования или др. кожных стигм (волосы, аплазия кожи, липома, гемангиома) требует исключения дизрафии спинного мозга. Простые крестцовые ямки не считаются признаком пороков развития спинного мозга. В этих случаях не следует проводить УЗИ позвоночника, поскольку большинство обнаруженных отклонений от нормы не будут иметь клинического значения. У детей грудного возраста <3 мес, которым требуется визуализация, УЗИ считается экономичным и неинвазивным методом. МРТ позвоночника — метод выбора для пациентов >3 мес, если есть веские подозрения на дизрафию спинного мозга.
б) Избыточная кожа. Рыхлые складки кожи необходимо отличать от врожденного дефекта эластической ткани или коллагена, такого как генерализованный эластолиз (синдром вялой кожи, cutis laxa), синдром Элерса-Данло (Ehlers-Danlos syndrome) или эластическая псевдоксантома. «Избыточная» кожа на задней поверхности шеи часто встречается при синдромах Тёрнера (Turner syndrome), Нунан (Noonan syndrome), Дауна (Down syndrome) и Клиппеля-Фейля (Klippel-Feil syndrome), а также при моносомии 1р36; более генерализованные складки кожи наблюдаются у новорожденных с трисомией по хромосоме 18 и карликовостью с короткими конечностями.
в) Амниотические перетяжки. Частичные или полные амниотические перетяжки, вызывающие дефекты конечностей и пальцев, встречаются у 1:10 000-45 000 в остальном здоровых детей. Амниотические перетяжки возникают в результате разрыва амниотической оболочки с последующим оплетением частей тела плода, особенно конечностей, сморщенными фиброзными амниотическими нитями. Это явление имеет спорадический характер с незначительным риском повторения. Формирование амниотических перетяжек обусловлено травмами живота во время беременности, амниоцентезом и наследственными дефектами коллагена, такими как синдром Элерса-Данло и несовершенный остеогенез. Лечение традиционно включает несколько хирургических процедур удлинения, таких как Z- и W-пластика.
В качестве альтернативы хирургическому вмешательству используется липоинъекция и множественные внутренние разрезы на глубокой поверхности перетяжки.
Адгезивные перетяжки затрагивают черепно-лицевую область и сопровождаются тяжелыми пороками развития, такими как энцефалоцеле и расщелины лица. Адгезивные перетяжки возникают в результате обширного срастания поврежденной ткани плода и неповрежденной амниотической оболочки. Черепно-лицевые дефекты вызваны не амниотическими перетяжками: они считаются результатом последовательного разрушения сосудов, иногда без сращения амниотической оболочки с тканями плода.
Комплекс конечности — стенки полостей тела обусловлен нарушением кровообращения на ранней стадии развития плода и затрагивает несколько эмбриональных структур. Он включает по крайней мере два из следующих трех пороков развития: экзэнцефалия или энцефалоцеле с расщелинами лица, незаращение грудной и/или брюшной стенки, а также дефекты конечностей.
г) Преаурикулярные синусы и ямки. Ямки и свищевые ходы, расположенные спереди от ушной раковины, м.б. результатом неполного сращения бугорков первой и второй жаберных дуг. Эти пороки развития бывают односторонними или двусторонними, иногда семейными, чаще встречаются у женщин, а также у афроамериканцев, а в некоторых случаях сопровождаются др. аномалиями ушей и лица. Преаурикулярные ямки наблюдаются при синдроме бранхио-ото-ренальной дисплазии I типа (ген EYA-1), АуД-заболевании, включающем пороки развития наружного уха, бранхиогенные свищи, глухоту и аномалии почек. При хронической инфекции свищей могут образовываться ретенционные кисты, которые периодически дренируются. В этих случаях часто требуется иссечение.
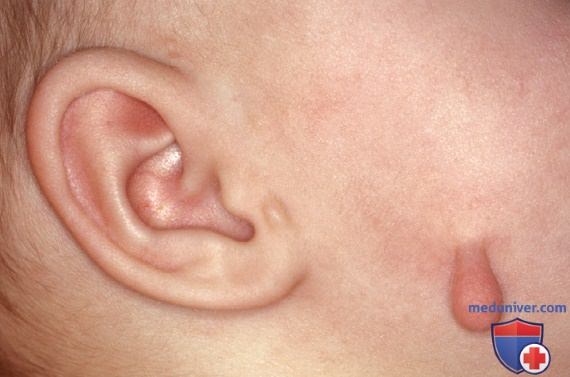
д) Добавочный козелок. Добавочный козелок выглядит как одиночная папула телесного цвета на ножке в преаурикулярной области кпереди от козелка. Реже добавочные козелки м.б. множественными или двусторонними и располагаются в преаурикулярной области, на щеке по линии нижней челюсти (рис. 1) или на латеральной стороне шеи кпереди от грудино-ключично-сосцевидной мышцы. В отличие от остальной части ушной раковины, которая развивается из второй жаберной дуги, козелок и добавочные козелки формируются из первой жаберной дуги. Добавочные козелки встречаются как изолированные дефекты или в рамках хромосомных синдромов первой жаберной дуги, включающих пороки развития ушей и лица, такие как расщелина верхней губы, расщелина нёба и гипоплазия нижней челюсти.
Рисунок 1. Добавочный козелок на щеке по линии челюсти
Добавочный козелок всегда обнаруживается при окулоаурикуловертебральном синдроме (синдром Гольденхара (Goldenhar syndrome)). Др. ассоциированные синдромы включают челюстно-лицевой дизостоз [синдром Тричера Коллинза (Treacher Collins syndrome), синдром Таунса-Брокса (Townes-Brocks syndrome), VACTERL (vertebral anomalies — аномалии позвоночника, anal atresia — атрезия ануса, cardiovascular anomalies — дефекты перегородок и др. пороки сердца, tracheoesophageal fistula — трахеопищеводный свищ с атрезией пищевода, renal defects — аномалии почек, limb defects — дефекты лучевой кости)] и синдром Вольфа-Хиршхорна (Wolf-Hirschhorn syndrome). Добавочный козелок удаляют хирургическим путем.
Исследования возможного повышения распространенности глухоты и аномалий МВП у пациентов с добавочными козелками и преаурикулярными ямками не дали однозначного результата. УЗИ почек следует проводить при обнаружении хотя бы одного из следующих признаков: глухота в семейном анамнезе, пороки развития ушной раковины и/или почек или гестационный диабет в анамнезе матери.
е) Бранхиогенные кисты и щитовидно-язычные кисты и свищи. Кисты и синусы на шее могут образовываться вдоль первой, второй, третьей или четвертой жаберной щели в результате неправильного закрытия во время эмбриональной жизни. Чаще всего наблюдаются кисты второй жаберной щели. Эти поражения м.б. односторонними или двусторонними (2-3%) и открываются на поверхность кожи или в глотку. Вторичная инфекция — показание для системной АБТ. Кисты и синусы шеи могут наследоваться АуД.
Щитовидно-язычные кисты и свищи — аналогичные дефекты, расположенные по средней линии шеи или около нее. Они могут простираться до основания языка. Патогномоничный признак — вертикальное смещение образования при глотании и высовывании языка. Почти у 50% пациентов детского возраста киста или свищ манифестируют в виде инфицированного объемного образования, расположенного по средней линии в верхней части шеи. Кисты в основании языка можно отличить от неопустившейся язычной ЩЖ с помощью сцинтиграфии. В отличие от бранхиогенных кист, киста щитовидно-язычного протока часто появляется после инфекции ВДП.
ж) Добавочные соски. Единичные или множественные добавочные соски м.б. односторонними или двусторонними и располагаются по линии, проходящей от передней подмышечной складки до паховой области. Они чаще встречаются у детей афроамериканского происхождения (3,5%), чем у детей европеоидной расы (0,6%). По данным литературы, распространенность составляет 0,1-0,99%. Добавочные соски могут иметь ареолы, но не всегда, и порой их можно ошибочно принять за врожденные невусы. Добавочные соски иссекают по косметическим причинам, но в остальном лечение не требуется. У детей с добавочными сосками в редких случаях обнаруживаются пороки развития почек или МВП, ЗНО, особенно рак МПС, и гематологические нарушения.
з) Ограниченные врожденные дефекты кожи и подкожной клетчатки или аплазия кожи (Aplasia cutis congenita). Области врожденного дефекта кожи обычно находятся на волосистой части головы в виде множественных или одиночных (70%) невоспалительных, четко очерченных, овальных или круглых язв размером 1-2 см (табл. 1). Внешний вид очагов зависит от времени их возникновения в период в/утробного развития. Дефекты, которые образуются на ранних сроках беременности, могут зажить до родов и будут выглядеть как атрофические фиброзные рубцы с сопутствующей алопецией, а более поздние дефекты имеют вид язв.
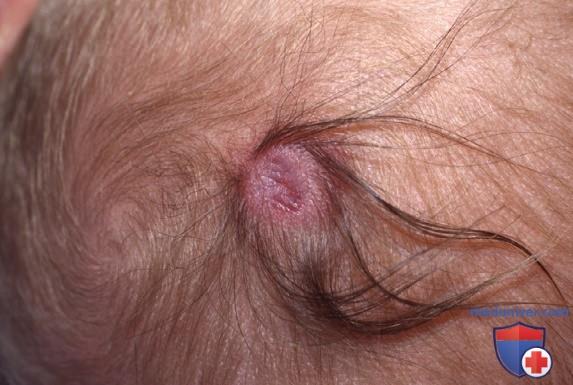
В большинстве случаев области врожденных дефектов кожи находятся на макушке, латеральнее средней линии, но аналогичные дефекты также могут появляться на лице, туловище и конечностях, где они часто симметричны и наблюдаются при в/утробной гибели второго плода из пары близнецов (fetus papyraceus, мумифицированный плод). Глубина и размер язвы различны: м.б. поражены только эпидермис и верхняя часть дермы, что приводит к минимальному рубцеванию или выпадению волос. Реже дефект распространяется на глубокие слои дермы, ПЖК и в отдельных случаях — на надкостницу, череп и твердую мозговую оболочку. Области аплазии кожи м.б. окружены венчиком волос — это известно как симптом волосяного венчика (рис. 2).
Рисунок 2. Единичный очаг врожденной аплазии кожи волосистой части головы на макушке, окруженный волосяным венчиком
Диагноз ставят на основании физикальных данных, указывающих на нарушение развития кожи в период в/ утробного развития. Иногда очаги аплазии кожи ошибочно принимают за повреждения от электродов на коже головы или родовую травму. Большинство случаев носит спорадический характер, но также встречаются АуД- и АуР-варианты аплазии кожи. Некоторые из них вызваны мутациями в гене BMS1, рибосомальной гуанозинтрифосфатазы.
У большинства детей с врожденной аплазией кожи др. пороков развития нет, но иногда это заболевание м.б. связано с отдельными физическими аномалиями или с синдромами пороков развития, включая синдромы Опица (Opitz syndrome), Адамса-Оливера (Adams-Oliver syndrome), окулоцереброкожный синдром, синдромы Йохансона-Близзарда (Johanson-Blizzard syndrome) и микроделеции 4р(-), Х-р22, трисомию 13-15 и дефекты хромосом 16-18 (см. табл. 1). Врожденная аплазия кожи также может обнаруживаться в сочетании с явной или скрытой эмбриологической аномалией, такой как ВПР легких, менингомиелоцеле, гастрошизис, омфалоцеле или дизрафия спинного мозга.
Врожденная аплазия кожи в сочетании с синдромом «исчезающего близнеца» (мумифицированный плод) вызвана ишемическими или тромботическими событиями в плаценте и сосудах плода, напр. гиповолемией, возникающей при резкой гемотрансфузии от выжившего близнеца к погибающему. Пузыри, или повышенная чувствительность кожи к повреждениям, и/или отсутствие или деформация ногтей в сочетании с врожденной аплазией кожи — хорошо известные проявления буллезного эпидермолиза.
Серьезные осложнения возникают редко и чаще связаны с большими звездчатыми очагами по средней линии теменной части головы. Описаны случаи кровотечения, вторичной местной инфекции и менингита. Если дефект небольшой, выздоровление протекает без осложнений, с постепенной эпителизацией и формированием атрофического рубца без волос в течение нескольких недель. Небольшие костные дефекты закрываются самостоятельно в 1-й год жизни. Большие или многочисленные дефекты кожи волосистой части головы могут потребовать пластики, но следует соблюдать осторожность, поскольку подлежащие патологические венозные структуры требуют сложных хирургических манипуляций.
Дефекты кожи туловища и конечностей, несмотря на большие размеры, рубцуются с формированием атрофических рубцов, которые впоследствии можно иссечь.
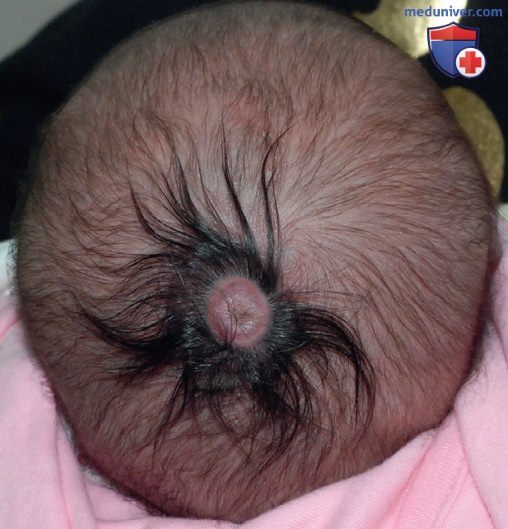
Признак волосяного венчика часто наблюдается при аплазии кожи, он также возможен при энцефалоцеле, менингоцеле, гетеротопии глиальных элементов или гамартоме. Для оценки этих поражений пациентам с признаком волосяного венчика без аплазии кожи часто показана МРТ ГМ (рис. 3).
Рисунок 3. Эластичный выступающий узелок без волос размером до 1,5 см с кольцом из темных толстых длинных волос, окружающих узелок и образующих «волосяной венчик»
и) Очаговые дисплазии кожи лица. Очаговые дисплазии кожи лица — редкая группа заболеваний, характеризующихся битемпоральными или преаурикулярными дефектами, которые напоминают рубцы или врожденную аплазию кожи. Очаговая дисплазия кожи лица 1-го типа (синдром Брауэра (Brauer)) наследуется по АуД-типу и обычно имеет легкие характерные изменения черт лица.
Очаговая дисплазия кожи лица 2-го типа (синдром Брауэра-Сетлейса (Brauer-Setleis syndrome)) и очаговая дисплазия кожи лица 3-го типа (синдром Сетлейса (Setleis syndrome)) характеризуются тонкой сморщенной кожей в периорбитальных областях, дистихиазом и/или отсутствием ресниц, монголоидным разрезом глаз, плоской переносицей, большими губами и складками кожи на лице. Очаговая дисплазия кожи лица 2-го типа наследуется по АуД-типу, а очаговая дисплазия кожи лица 3-го типа считается АуР-заболеванием, связанным с мутациями в TWIST2.
Описаны АуД-случаи очаговой дисплазии кожи лица 3-го типа, которые обусловлены дупликацией/утроением хромосомной области 1р36.22р36.21. Очаговая дисплазия кожи лица 4-го типа не имеет др. кожных изменений, наследуется как по АуД, так и по АуР-типу и обусловлена мутациями в CYP26C1.
к) Очаговая гипоплазия дермы (синдром Гольца-Горлина). Редкое врожденное мезоэктодермальное и эктодермальное заболевание, очаговая гипоплазия дермы, характеризуется дисплазией соединительной ткани в коже и костях. Синдром Гольца-Горлина (Goltz-Gorlin syndrome) — Х-сцепленное доминантное заболевание, вызванное мутациями в гене PORCN, проявляется в виде многочисленных папиллом светло-коричневого цвета. Др. кожные изменения включают линейные атрофические поражения; сетчатую гипопигментацию и гиперпигментацию; телеангиэктазии; врожденное отсутствие кожи; ангиофибромы в виде бородавчатых элементов; папилломы губ, языка, периоральной области, вульвы, ануса, а также паховой, подмышечной и околопупочной области. Очаговая алопеция, нарушение потоотделения и дистрофия ногтей — дополнительные, менее частые эктодермальные патологии.
Самые частые пороки развития скелета — синдактилия, клинодактилия, полидактилия и сколиоз. Остеопатия полосчатая — тонкие параллельные вертикальные полосы на рентгенограммах в метафизах длинных костей у пациентов с этим заболеванием. Они очень характерны для очаговой гипоплазии дермы, но не считаются патогномоничными. Также типичным признаком будут многие патологии глаз, чаще всего — колобомы, косоглазие, нистагм и микрофтальм. Часто отмечается небольшой рост, гипоплазия эмали, аномалии мягких тканей и своеобразные папиллярные узоры. Иногда возможны когнитивные нарушения. Специфического лечения нет.
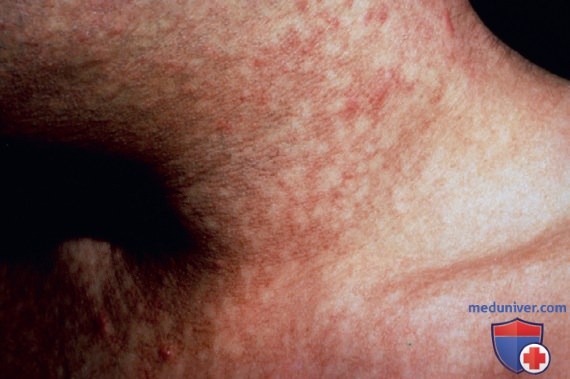
л) Врожденный дискератоз (синдром Цинссера-Энгмена-Коула). Врожденный дискератоз (синдром Цинссера-Энгмена-Коула (Zinsser-Cole-Engmann syndrome)), редкий семейный синдром, классически состоит из триады — сетчатая гиперпигментации кожи (рис. 4), дистрофия ногтей и лейкоплакия слизистой оболочки в сочетании с иммунологическими и гематологическими нарушениями. У пациентов с врожденным дискератозом также наблюдаются признаки преждевременного старения и возрастание частоты случаев злокачественных опухолей, особенно плоскоклеточного рака.
Рисунок 4. Сетчатая диспигментация на шее пациента с врожденным дискератозом
Врожденный дискератоз м.б. Х-сцепленным рецессивным (ген DKC-1), АуД-(гены hTERC и TINF2) или АуР- (ген N0LA3) заболеванием. Врожденный дискератоз дебютирует в детстве, чаще всего как дистрофия ногтей. Ногти становятся атрофичными и ребристыми в продольном направлении с прогрессированием до птеригиума и полной потери ногтей. Кожные изменения появляются уже после изменений ногтей и включают сетчатую серо-коричневую пигментацию, атрофию и телеангиэктазии, особенно на шее, лице и груди. Также характерны гипергидроз и гиперкератоз ладоней и подошв, редкие волосы на коже головы и незначительное образование пузырей на руках и ногах. Иногда наблюдается блефарит, эктропион и чрезмерное слезотечение из-за атрезии слезных протоков. Лейкокератоз слизистой полости рта способен трансформироваться в плоскоклеточный рак.
Могут поражаться и др. слизистые оболочки, включая конъюнктиву, уретру и половые органы. Часто встречаются инфекции, ЗНО, легочный фиброз и недостаточность костного мозга, большинство пациентов умирают в возрасте до 40 лет. Эффективного лечения нет. Трансплантация аллогенных гемопоэтических стволовых клеток позволяет излечить пациента при недостаточности костного мозга.
м) Складчатая пахидермия (Cutis verticis gyrata). Cutis verticis gyrata (складчатая пахидермия) — редкое изменение кожи головы, которое чаще встречается у мужчин, может присутствовать с рождения или развиваться в подростковом возрасте. На коже волосистой части головы видны извитые приподнятые складки толщиной 1-2 см, обычно на лобно-затылочной оси. В отличие от дряблой кожи при др. заболеваниях, эти складки не исчезают при растягивании. Первичная складчатая пахидермия м.б. связана с умственной отсталостью, пигментным ретинитом, нейросенсорной тугоухостью и аплазией ЩЖ. Вторичная складчатая пахидермия часто сопровождается хроническими воспалительными заболеваниями, опухолями, невусами и акромегалией.