В большинстве случаев СД у детей подразделяется на две большие группы: СД-1 и СД-2, однако 1-10% случаев вызваны моногенными нарушениями — наследственными дефектами функции β-клеток и работы инсулина, редкими формами митохондриального СД.
а) Генетические дефекты функции β-клеток:
1. Транзиторный неонатальный сахарный диабет. Неонатальный СД является транзиторным в ~50% случаев, однако в период нормальной толерантности к глюкозе у 50-60% этих пациентов развивается перманентный СД (в среднем — в 14 лет). Есть сведения о пациентах с классическим СД-1, у которых в периоде новорожденности был транзиторный СД.
Остается неясным, существует ли причинно-следственная связь между транзиторным диабетом в периоде новорожденности и классическим СД-1 в дальнейшей жизни или это случайность (рис. 1).
Рисунок 1. Генетическая диагностика определяет тактику лечения. Схематическое изображение генетических причин неонатального диабета и роль генетической диагностики
Симптомы транзиторного СД у новорожденного появляются в первую неделю жизни и сохраняются от нескольких недель до нескольких месяцев вплоть до спонтанного исчезновения (средняя продолжительность — 12 нед). Транзиторный СД чаще встречается у младенцев и детей с низкой МТ для своего гестационного возраста. Характерные признаки: гипергликемия и выраженная глюкозурия с последующими тяжелым обезвоживанием и иногда метаболическим ацидозом, но с минимальными кетонемией и/или кетонурией, или полным их отсутствием. У пациентов могут быть обнаружены пупочная грыжа или макроглоссия.
Инсулиновая реакция на глюкозу или толбутамид, как правило, низкая или вовсе отсутствует; базальная концентрация инсулина плазмы в норме. После спонтанного выздоровления инсулиновая реакция на эти же стимулы восстанавливается до нормальной, что свидетельствует о функциональной задержке созревания β-клеток со спонтанным разрешением. Сообщалось о наличии этого синдрома у родных братьев и сестер. Около 70% случаев обусловлены аномалиями импринтингового локуса на хромосоме 6q24, что приводит к сверхэкспрессии генов отцовского происхождения — гена плеоморфной аденомы (PLAGL1/ZAC) и хорионаденомы (HYMAI).
В большинстве остальных случаев транзиторный диабет обусловлен мутациями в АТФ-зависимых калиевых каналах: при этом мутации в генах АТФ-зависимых калиевых каналов в большинстве случаев приводят к перманентному неонатальному СД, однако мутации, ведущие к транзиторному неонатальному СД, и мутации, вызывающие перманентный неонатальный СД, практически не пересекаются. Транзиторный неонатальный СД следует отличать от тяжелой гипергликемии вследствие тяжелой гипертонической дегидратации: она обычно возникает у младенцев после периода новорожденности и быстро реагирует на регидратацию с минимальной потребностью в инсулине или вообще без нее.
Введение инсулина обязательно в активную фазу СД у новорожденного: сначала необходимы регидратация и в/в-введение инсулина; переход на п/к-введение инсулина осуществляют после достижения клинически стабильного состояния. Успешно применяют разнообразные режимы, в т.ч. введение инсулина средней продолжительности и инсулина длительного действия за 1-2 введения/сут, или постоянную инсулинотерапию с помощью инсулиновой помпы.
Начальная доза — 1-2 ЕД/кг/сут; ее следует корректировать в зависимости от уровня глюкозы крови. В случае рецидивирующей гипогликемии либо после достижения возраста 2 мес возможно постепенное снижение дозы инсулина. В настоящее время доступен и рекомендуется к проведению всем пациентам генетический анализ на аномалии 6q24 хромосомы и на дефекты АТФ-зависимых калиевых каналов (также рекомендуется медико-генетическое консультирование для оценки риска рецидива).
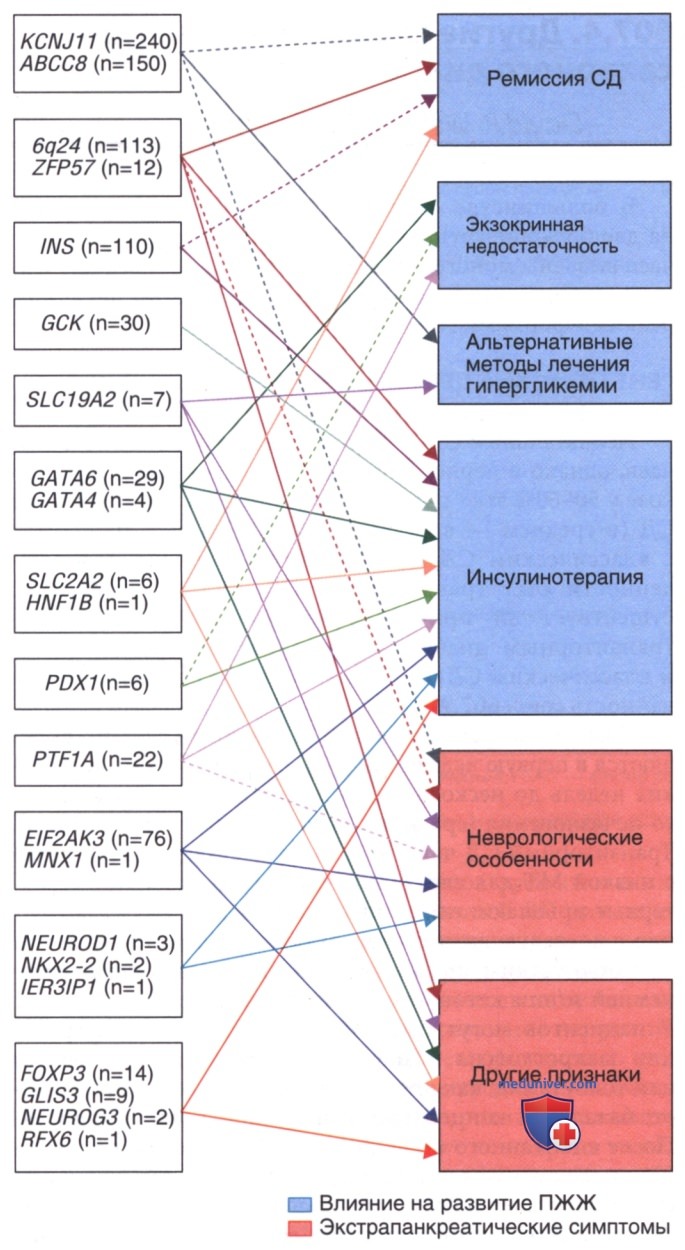
2. Перманентный неонатальный сахарный диабет. Перманентный СД у новорожденных в 50% случаев обусловлен мутациями в генах KCNJ11 (калиевый канал внутреннего выпрямления J, элемент 11) и АВСС8 (АТФ-связывающий кассетный транспортер, подсемейство С, элемент 8) (см. рис. 1 и рис. 2). Эти гены кодируют субъединицы Kir6.2 и SUR1 аденозинтрифосфат-чувствительного калиевого канала, участвующего в основном этапе секреции инсулина β-клетками. Некоторые случаи обусловлены агенезией ПЖЖ в результате гомозиготных мутаций в гене IPF-1 (гетерозиготные мутации приводят к MODY4), гомозиготных мутаций в гене глюкокиназы (гетерозиготные мутации приводят к MODY2) и мутаций в гене инсулина.
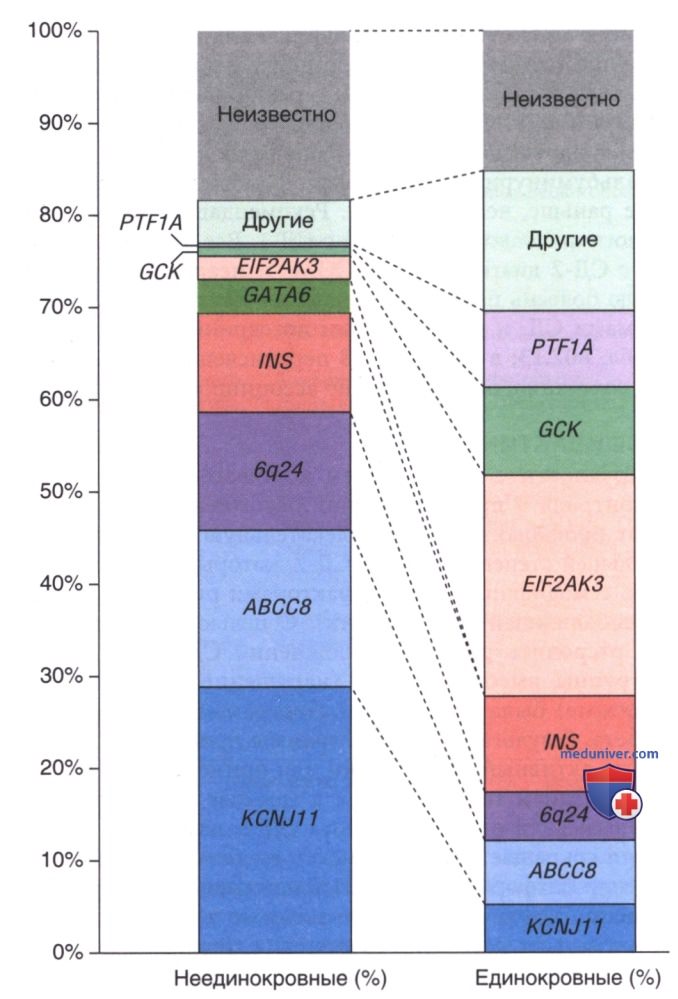
Рисунок 2. Различные генетические причины неонатального сахарного диабета у пациентов, рожденных от родителей, не являющихся кровными родственниками (n = 790), и близкородственных родителей (n = 230). Кровное родство: родители являются троюродными или более близкими родственниками, или общая гомозиготность >1-56%. Генетические мутации у менее чем 2-5% пациентов из обеих групп были отнесены к другой категории.
Почти все эти младенцы рождаются с низкой МТ, т.к. инсулин играет роль фактора в/утробного роста. Известны случаи данной патологии у близнецов или у более чем у одного младенца в семье. Младенцы с перманентным неонатальным СД могут изначально иметь эугликемию: патология проявляется в период между рождением и 6 мес жизни (средний возраст — 5 нед). Степень тяжести варьирует: у 20% пациентов есть неврологические особенности; для пациентов с заболеванием тяжелой степени характерны задержка развития, эпилепсия и неонатальный диабет (синдром DEND). Менее тяжелые формы DEND обозначают как DEND промежуточного типа или i-DEND.
Активирующие мутации в гене KCNJ11 (кодирование субъединицы Kir6.2 АТФ-чувствительного калиевого канала) сопряжены с транзиторным неонатальным СД и с перманентным неонатальным СД, при этом каждая мутация связана с определенным фенотипом. Более 90% таких пациентов реагируют на препараты сульфонилмочевины (в более высоких дозах, чем при лечении СД-2), однако пациенты с тяжелым неврологическим заболеванием могут реагировать на терапию в меньшей степени.
Предполагалось, что мутации в гене АВСС8 (кодирует субъединицу SUR1 калиевого канала) в меньшей степени реагируют на действие сульфонилмочевины (т.к. эта субъединица связывает препараты сульфонилмочевины), однако все же известны некоторые мутации, при которых пациенты были успешно переведены с инсулинотерапии на пероральную терапию. Существует несколько протоколов перевода с инсулина на глибенкламид: обычно удается добиться стабилизации состояния на дозах 0,4-1 мг/кг/сут. Всем пациентам с СД, диагностированным до 6 мес (и, возможно, до 12 мес), рекомендуется генетическое обследование, т.к. ~50% пациентов с неонатальным диабетом имеют мутации К-канала и могут быть переведены на терапию сульфонилмочевиной с резким улучшением гликемического контроля и качества жизни.
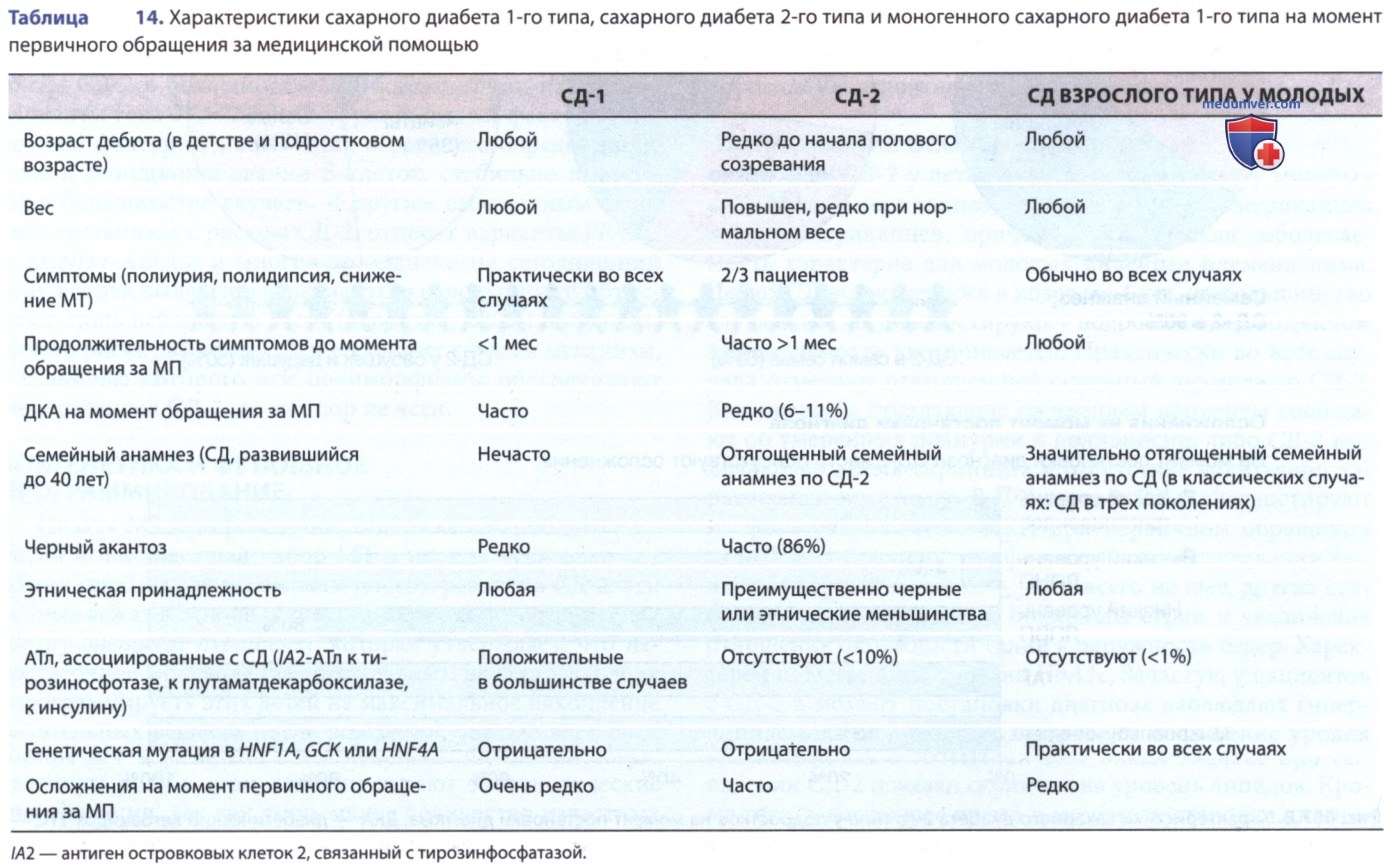
3. Диабет взрослого типа у молодых. Некоторые формы СД связаны с моногенными дефектами функции β-клеток. До идентификации генетических дефектов диагноз СД ставили на основании клинических симптомов и описывали термином «MODY». Данный подтип СД состоит из группы гетерогенных клинических синдромов с дебютом до 25 лет, АуД-типом наследования и первичным дефектом секреции инсулина. Строгие критерии диагностики MODY: СД по крайней мере в трех поколениях с АуД-типом наследования и его дебют до 25 лет как минимум у одного члена семьи. Мутации были обнаружены по меньшей мере в 11 различных генах, что объясняет доминантно-наследуемые моногенные дефекты секреции инсулина, для обозначения которых используется термин «MODY».
Американская ассоциация диабетологов объединяет эти расстройства в более широкую группу генетических дефектов функции β-клеток: как правило, 11 из этих нарушений соответствуют клиническим критериям MODY (перечислены в табл. 19); только три из них (MODY2, MODY3 и MODY5) составляют 90% случаев данной категории среди европейского населения, в то время как в других этнических группах распределение может отличаться. Все остальные формы обусловлены генетическими дефектами различных факторов транскрипции — за исключением MODY2, который вызван мутациями в гене глюкокиназы (см. табл. 19).
- Диабет взрослого типа у молодых 2-го типа. Это вторая по распространенности форма MODY — 15-30% всех пациентов с диагнозом MODY. Глюкокиназа играет важную роль в распознании глюкозы β-клетками, а гетерозиготные мутации в причинном гене приводят к умеренному снижению реакции β-клеток ПЖЖ на глюкозу. Гомозиготы с подобными мутациями абсолютно неспособны к секреции инсулина в ответ на повышение глюкозы и приводят к одной из форм перманентного неонатального СД. Пациенты с гетерозиготными мутациями имеют более высокий порог высвобождения инсулина, но способны адекватно секретировать инсулин при более высоких уровнях глюкозы крови [>125 мг/дл (>7 ммоль/л)]: это приводит к относительно легкой форме диабета (HbA1c обычно <7%) с легкой гипергликемией натощак и нарушением толерантности к глюкозе у большинства пациентов.
MODY2 может быть ошибочно диагностирован как СД-1 у детей младшего возраста, как гестационный СД у беременных или как хорошо контролируемый СД-2 у взрослых (см. табл. 14). Точный диагноз играет важную роль, т.к. в большинстве случаев заболевание не прогрессирует и может не требовать лечения (кроме гестационного СД). Возможно назначение небольших доз инсулина PRN. Лечение пероральными ЛП (сульфонилмочевиной и аналогичными) может быть успешным и более приемлемым для многих пациентов.
- Диабет взрослого типа у молодых 3-го типа. У пациентов с мутациями в генах ядерного фактора-1α гепатоцитов наблюдается нарушение углеводного обмена от нарушения толерантности к глюкозе до тяжелой формы СД и склонность к развитию сосудистых осложнений. Это наиболее распространенный тип MODY: 50-65% всех случаев. Такие пациенты очень чувствительны к действию сульфонилмочевины: обычно эффективными оказываются относительно низкие дозы пероральных ЛС (по крайней мере на ранних стадиях заболевания). У детей данную форму MODY иногда ошибочно диагностируют как СД-1 и лечат инсулином. Оценка аутоиммунных маркеров помогает исключить СД-1: в настоящее время генетический анализ доступен и показан пациентам с относительно легкой формой СД и семейным анамнезом, указывающим на АуД-тип наследования. Диагностика может помочь избежать ненужного лечения инсулином и специального генетического консультирования.
- Менее распространенные формы моногенного диабета. Ядерный фактор гепатоцитов-4α (MOODY1), фактор промотора инсулина (IPF-1, также известный как PDX-1) (MODY4), ядерный фактор гепатоцитов-10/TCF2 (MODY5) и NeuroD1 (MODY6) — участвующие в развитии и функционировании β-клеток транскрипционные факторы, мутации в которых приводят к различным редким формам MODY. Помимо СД эти мутации могут иметь определенные особенности, не связанные с гипергликемией: напр., M0DY1 ассоциирован с низким уровнем триглицеридов и липопротеинов, a M0DY5 — с кистами и нарушением функции почек. С точки зрения лечения MODY1 и M0DY4 могут реагировать на препараты сульфонилмочевины, в то время как MODY5 не отвечает на пероральные препараты и требует лечения инсулином. Дефекты NeuroDl встречаются крайне редко: об их клинических признаках и особенностях течения известно очень мало.
Первичные или вторичные дефекты в транспортере глюкозы-2 (инсулиннезависимый транспортер глюкозы) также могут быть связаны с развитием СД. СД может быть следствием полиморфизма гена гликогенсинтазы: этот фермент имеет ключевое значение в накоплении глюкозы в виде гликогена в мышцах; пациенты с таким дефектом имеют выраженную инсулинорезистентность и АГ, семейный анамнез СД. Синдром Вольфрама — другая форма инсулинзависимого СД — характеризуется сочетанием несахарного диабета, СД, атрофии зрительного нерва и глухоты (для его обозначения используют акроним DIDMOAD). Некоторые пациенты с СД имеют выраженную недостаточность инсулина, в то время как у других она сохранена, о чем судят по уровню С-пептида. По некоторым оценкам, распространенность DIDMOAD — 1:770 000 живорожденных детей.
Последовательность появления составляющих синдрома следующая: неаутоиммунный инсулинзависимый СД в первые 10 лет жизни; несахарный диабет центрального генеза и нейросенсорная тугоухость у 2/3-2/4 пациентов во 2-м десятилетии; аномалии МПС у ~50% пациентов в 3-м десятилетии; неврологические осложнения (мозжечковая атаксия, миоклональная эпилепсия) у 1/2-2/3 пациентов на 4-м десятилетии жизни. Другие особенности — первичная атрофия гонад у большинства мужчин и прогрессирующее нейродегенеративное заболевание с летальным исходом в результате нейрореспираторного приступа (средний возраст — 30 лет). Некоторые (но не все) случаи вызваны мутациями в гене WFS1 (вольфрамин) на хромосоме 4р.
Синдром Вольфрама 2-го типа характеризуется ранним появлением атрофии зрительного нерва, СД, нейросенсорной тугоухостью и низкой продолжительностью жизни, а несахарный диабет, сопряженный с мутациями в гене CISD2, отсутствует. Другие формы синдрома Вольфрама могут быть обусловлены мутациями в митохондриальной ДНК.
- Митохондриальные генные дефекты. Точечные мутации в митохондриальной ДНК ассоциированы с наследуемым по материнской линии СД и глухотой. Наиболее распространенная мутация митохондриальной ДНК — точечная мутация m.3243A>G в гене лейцина транспортной РНК. Эта мутация идентична мутации при MELAS-синдроме (миопатия, энцефалопатия, лактат-ацидоз и инсультоподобные эпизоды), однако данный синдром не связан с СД, а фенотипическая экспрессия одного и того же дефекта может различаться. СД в большинстве этих случаев проявляется скрыто, но у 20% пациентов наблюдается острая форма, напоминающая СД-1. Средний возраст диагностики — 37 лет, однако известны случаи диагностики в возрасте 11 лет, при этом не все пациенты страдают глухотой.
Эта мутация присутствует у 1,5% японцев, больных СД, что выше, чем распространенность в других этнических группах. Следует избегать применения метформина у таких пациентов в связи с потенциальным риском развития тяжелой формы лактат-ацидоза на фоне митохондриальной дисфункции. У некоторых детей с мутациями митохондриальной ДНК, влияющими на комплекс I и/или комплекс IV, также может развиться СД.
4. Аномалии гена инсулина. СД различной степени тяжести может быть следствием мутаций в гене инсулина, снижающих активность инсулина на рецепторном уровне. Дефекты генов инсулина чрезвычайно редки и могут быть сопряжены с относительно легкими формами диабета или даже с нормальной толерантностью к глюкозе. СД может также развиться у пациентов с нарушением превращения проинсулина в инсулин (АуД-дефект). Такие дефекты отличаются высокой концентрацией инсулина по данным радиоиммунологического анализа, в то время как для MODY и дефектов глюкозного транспортера-2 характерны относительный или абсолютный дефицит секреции инсулина.
б) Генетические дефекты действия инсулина. Различные генетические мутации в рецепторе инсулина могут ослаблять действие инсулина на уровне рецепторов или нарушать пострецепторную сигнальную функцию, приводя к инсулинорезистентности. Самая легкая форма синдрома с мутациями в рецепторе инсулина раньше была известна как инсулинорезистентность типа А: патология сопряжена с гирсуртизмом, гиперандрогенией и кистами яичников у женщин; ожирение не характерно; может присутствовать папиллярно-пигментная дистрофия кожи; средняя продолжительность жизни существенно не изменяется. Более тяжелые формы инсулинорезистентности наблюдаются при двух мутациях в гене рецептора инсулина, вызывающих синдром Донохью (ранее известный как лепречаунизм) и синдром Рабсона-Менденхолла у детей.
1. Синдром Донохью. Характерные признаки: задержка в/утробного развития, гипогликемия натощак и постпрандиальная гипергликемия, выраженная инсулиновая резистентность. Тяжелую гиперинсулинемию подтверждает пероральный тест на толерантность к глюкозе. Описывают различные дефекты рецептора инсулина, что говорит о важной роли инсулина и его рецептора для развития плода и, возможно, для морфогенеза. Многие такие пациенты умирают на первом году жизни. Потенциальные методы лечения включают в себя высокие дозы инсулина, метформин и непрерывную подачу инсулиноподобного фактора роста-1 через инсулиновую помпу.
2. Синдром Рабсона-Менденхолла. Данную нозологическую единицу определяют как промежуточные клинические проявления между папиллярно-пигментной дистрофией кожи с инсулинорезистентностью типа А и синдромом Донохью. Особенности синдрома: уже упомянутые выраженная инсулинорезистентность и папиллярно-пигментная дистрофия кожи, аномалии зубов и ногтей и гиперплазия шишковидной железы. Неясно, полностью ли отличается этот синдром от синдрома Донохью; однако при сравнении отмечено, что пациенты с синдромом Рабсона-Менденхолла обычно живут существенно дольше. Методы лечения, приносящие умеренную пользу: препараты инсулиноподобного фактора роста 1 и лептина.
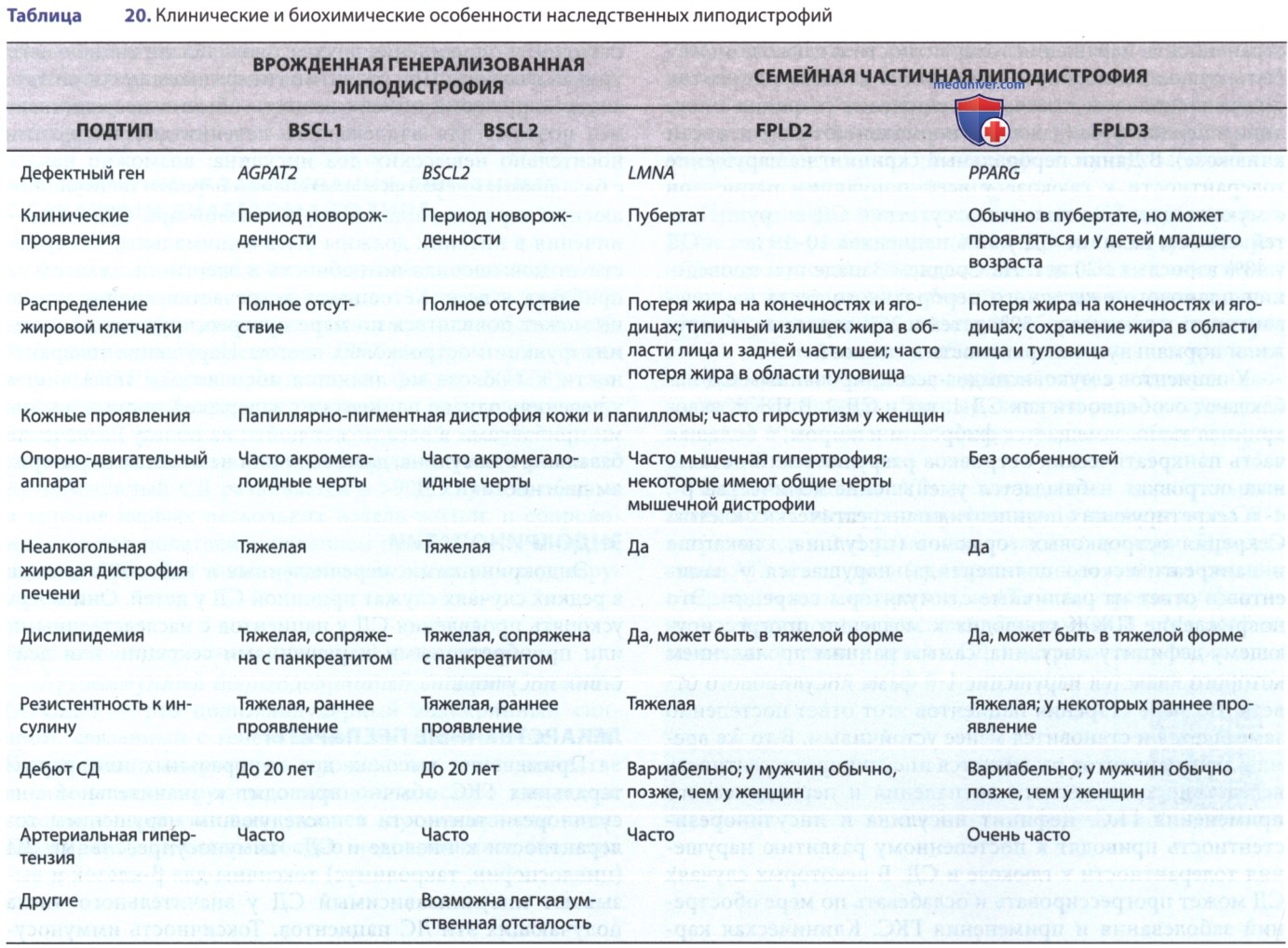
3. Липоатрофический сахарный диабет. Различные формы липодистрофий сопряжены с инсулинорезистентностью и СД (табл. 20). Семейная частичная липоатрофия, или липодистрофия, сопряжена с мутациями в гене LMNA, кодирующем белки ядерной оболочки — ламин А и С. Тяжелая врожденная генерализованная липоатрофия сопряжена с мутациями в генах серпина и AGPAT2, однако механизм, посредством которого эти мутации приводят к инсулиновой резистентности и СД, неизвестен.
4. Синдром мышечной скованности. Это очень редкое аутоиммунное нарушение ЦНС, для которого характерны прогрессирующая скованность, болезненные спазмы аксиальных мышц и крайне высокие титры АТл к глутаматдекарбоксилазе. Примерно у 1/3 пациентов развивается СД-1.
5. Системная красная волчанка. В редких случаях у пациентов с СКВ могут развиваться аутоАТл к рецепторам инсулина, что приводит к инсулинорезистентности и СД.
в) Муковисцидоз-ассоциированный диабет. В связи с увеличением продолжительности жизни у пациентов с муковисцидозом все большему числу из них диагностируют муковисцидоз-ассоциированный СД: женщины в большей степени подвержены такому риску, а распространенность заболевания повышается с возрастом до достижения 40 лет (после 40 лет распространенность снижается — предположительно в связи с тем, что только пациенты с легкой формой муковисцидоза выживают после этого возраста).
Отмечают взаимосвязь патологии с недостаточностью ПЖЖ, т.о., пациенты с мутациями муковисцидозного трансмембранного регулятора проводимости классов I и II могут быть подвержены большему риску. По данным крупного многоцентрового исследования в США, распространенность заболевания составляет 17% среди женщин и 12% среди мужчин (всех возрастов). Межгрупповые исследования показывают, что распространенность нарушения толерантности к глюкозе может быть существенно более высокой, и у 65% пациентов с муковисцидозом отмечается снижение секреции инсулина в первой фазе (даже при нормальной толерантности к глюкозе).
В Дании пероральный скрининг на нарушение толерантности к глюкозе у всей популяции пациентов с муковисцидозом показал отсутствие СД в группе детей <10 лет, наличие СД у 12% пациентов 10-19 лет и СД у 48% взрослых >20 лет. На Среднем Западе при проведении планового ежегодного перорального теста на толерантность к глюкозе у 50% детей и 25% взрослых обнаружили нормальную толерантность к глюкозе.
У пациентов с муковисцидоз-ассоциированным СД наблюдают особенности как СД-1, так и СД-2. В ПЖЖ экзокринная ткань замещается фиброзом и жиром, и большая часть панкреатических островков разрушается. В остальных островках наблюдается уменьшение количества β-, α- и секретирующих полипептид панкреатических клеток. Секреция островковых гормонов (инсулина, глюкагона и панкреатического полипептида) нарушается у пациентов в ответ на различные стимуляторы секреции.
Это повреждение ПЖЖ приводит к медленно прогрессирующему дефициту инсулина, самым ранним проявлением которого является нарушение 1-й фазы инсулинового ответа. По мере старения пациентов этот ответ постепенно замедляется и становится менее устойчивым. В то же время у этих пациентов развивается инсулинорезистентность вследствие хронического воспаления и периодического применения ГКС. Дефицит инсулина и инсулинорезистентность приводят к постепенному развитию нарушения толерантности к глюкозе и СД. В некоторых случаях СД может прогрессировать и ослабевать по мере обострений заболевания и применения ГКС. Клиническая картина сходна с таковой при СД-2: заболевание протекает скрыто, а ДКА возникает редко. Титры островковых АТл отрицательные. Микрососудистые осложнения развиваются медленнее, чем при типичных СД-1 и СД-2.
Макро-сосудистые осложнения не успевают развиться вследствие короткой продолжительности жизни этих пациентов. На возникновение и течение СД влияют несколько факторов, присущих только муковисцидозу, напр.: частые инфекции, связанные с повышением и снижением резистентности к инсулину; увеличение энергетической потребности в связи с инфекцией и заболеванием легких; проявление мальабсорбции, несмотря на применение ферментных препаратов; замедление всасывания питательных веществ вследствие снижения скорости транзита пищи по кишечнику; наличие заболеваний печени (часто); анорексия и тошнота (часто); широкий диапазон ежедневного количества потребляемой пищи в зависимости от состояния здоровья пациента; нарушения секреции инсулина и глюкагона (в отличие от аутоиммунного диабета, при котором нарушена только секреция инсулина).
Нарушение толерантности к глюкозе и муковисцидоз-ассоциированный СД сопряжены с плохими прибавками МТ. Есть данные о том, что лечение инсулином способствует набору веса и замедляет ухудшение состояния легких. Исходя из этих наблюдений, руководства Фонда муковисцидоза, Американской диабетической ассоциации, Общества детских эндокринологов рекомендуют проведение обследования для исключения СД у всех детей с муковисцидозом, начиная с возраста 10 лет. Несмотря на споры относительно идеального метода исследования, в настоящее время рекомендуют проводить двухчасовой тест на толерантность к глюкозе, хотя возможно, что достаточно и определения двухчасового постпрандиального уровня глюкозы.
При развитии гипергликемии сопутствующие нарушения обмена веществ обычно несущественны, поэтому для надлежащего лечения достаточно относительно невысоких доз инсулина: возможно начать с базального инсулина и постепенно перейти на базис-болюсную терапию, аналогичную таковой при СД-1. Ограничения в питании должны быть минимальны, т.к. существует повышенная потребность в энергии и желательна прибавка в весе. Кетоацидоз встречается крайне редко, но может появляться по мере прогрессирующего снижения функции островковых клеток.
Нарушение толерантности к глюкозе не является абсолютным показанием к лечению, однако пациентам с задержкой роста и низкими прибавками в весе может пойти на пользу назначение базального инсулина, даже если они не отвечают критериям диагностики СД.
г) Эндокринопатии. Эндокринопатии, перечисленные в табл. 1, лишь в редких случаях служат причиной СД у детей. Они могут ускорять проявления СД у пациентов с наследственными или приобретенными нарушениями секреции или действия инсулина.
д) Лекарственные препараты. Применение высоких доз пероральных или парентеральных ГКС обычно приводит к значительной инсулинорезистентности с последующим нарушением толерантности к глюкозе и СД. Иммуносупрессивные ЛП (циклоспорин, такролимус) токсичны для β-клеток и вызывают инсулинзависимый СД у значительного числа получающих эти ЛС пациентов. Токсичность иммуносупрессивных ЛП для β-клеток ПЖЖ была одним из факторов, ограничивающих целесообразность их применения для подавления происходящего аутоиммунного разрушения β-клеток. Стрептозотоцин и родентицид «Вакор» также токсичны для β-клеток и приводят к СД.
Не существует единых рекомендаций лечения стероид-индуцированной гипергликемии у детей. У многих пациентов, получающих ГКС в высоких дозах, наблюдают повышенные уровни глюкозы крови в течение дня и вечером и нормогликемию поздно вечером или натощак. Как правило, значительную гипергликемию в стационарных условиях лечат инсулином короткого действия PRN. Базальный инсулин может быть добавлен при значимой гипергликемии натощак. Лечение в амбулаторных условиях может быть более сложным, однако PRN используют схемы базис-болюсных режимов, применяемых при СД-1.
е) Генетические синдромы, сопряженные с сахарным диабетом. Существует ряд редких генетических синдромов, сопряженных с инсулинзависимым СД или нарушением толерантности к глюкозе (см. табл. 1). Такие синдромы представляют собой широкий спектр заболеваний — от преждевременного старения клеток, как при синдромах Вернера и Коккейна, до морбидного ожирения, сопряженного с гиперинсулинизмом, инсулинорезистентностью и нарушением толерантности к глюкозе, как при синдроме Прадера-Вилли. Некоторые из этих синдромов характеризуются первичными нарушениями в рецепторе инсулина или в АТл к рецептору инсулина без какого-либо нарушения его секреции.
Несмотря на редкость, эти синдромы уникальны для понимания многочисленных причин нарушения углеводного обмена вследствие дефектной секреции инсулина или действия инсулина на клеточном рецепторном или пострецепторном уровне.
ж) Аутоиммунные заболевания, связанные с сахарным диабетом 1-го типа:
1. IPEX-синдром. Х-сцепленный синдром иммунной дисрегуляции, полиэндокринопатии и энтеропатии — генетический синдром, ведущий к развитию аутоиммунных заболеваний. У большинства пациентов с IPEX-синдромом мутации в гене FOXP3 (транскрипционный фактор FOXP3), специфическом маркере естественных и адаптивных регуляторных Т-клеток, приводят к тяжелой иммунной дисрегуляции и неконтролируемой аутоиммунной реакции. Аутоиммунный СД развивается в >90% случаев, обычно в течение первых нескольких недель жизни, и сопровождается энтеропатией, снижением прибавки МТ и отставанием в росте, а также другими аутоиммунными нарушениями.
2. Аутоиммунные полиэндокринные синдромы. Аутоиммунный полиэндокринный синдром 1-го типа (APCED) — это полигландулярный эндокринный синдром, связанный с генетической мутацией в гене AIRE. Как правило, впервые он проявляется в младенчестве в виде рецидивирующего кожно-слизистого кандидоза с последующей гипокальциемией (аутоиммунный гипопаратиреоз), недостаточностью надпочечников (болезнь Аддисона), СД-1, гипотиреозом (болезнь Хасимото), целиакией и другими аутоиммунными расстройствами.
Более распространен аутоиммунный полиэндокринный синдром 2-го типа, который обычно представляет собой болезнь Аддисона в сочетании с другим аутоиммунным заболеванием либо сочетание двух любых аутоиммунных заболеваний. Каждый пациент с аутоиммунным заболеванием имеет высокий риск развития СД-1 (равно как и каждый пациент с СД-1 подвержен повышенному риску другого аутоиммунного заболевания) и нуждается в консультировании относительно признаков впервые выявленного СД. В табл. 13 представлены рекомендации относительно диагностических тестов для выявления других аутоиммунных заболеваний у пациентов с СД-1.
Хронический лимфоцитарный тиреоидит (тиреоидит Хасимото) у детей часто сопряжен с СД-1. Примерно 20% пациентов с инсулинозависимым СД имеют тиреоидные АТл в сыворотке — их распространенность в 2-20 раз выше, чем в контрольных популяциях. Лишь у небольшой доли данных пациентов развивается клинический гипотиреоз.
Время между диагностикой СД и заболеванием ЩЖ в среднем ~5 лет. Целиакия вследствие повышенной чувствительности к пищевому глютену — еще одно аутоиммунное заболевание, часто встречающееся у детей с СД-1. Согласно оценкам, у 7-15% детей с СД-1 целиакия развивается в течение 1-6 лет с момента диагностики СД, и распространенность ее существенно выше у детей <4 лет и у женщин.
У детей младшего возраста с СД-1 и целиакией могут отмечаться расстройства ЖКТ (спазмы в животе, диарея, запоры, ГЭР), отставание в росте как следствие недостаточных весовых прибавок, необъяснимые гипогликемические состояния вследствие нарушения всасывания питательных веществ и, реже, гипокальциемия в связи с тяжелой мальабсорбцией витамина D; в некоторых случаях заболевание может протекать бессимптомно.
При одновременном существовании СД и заболевания ЩЖ следует рассмотреть также вероятность аутоиммунной недостаточности надпочечников. На нее могут указывать: снижение потребности в инсулине, повышенная пигментация кожи и слизистой оболочки щек, пристрастие к соленой пище, слабость, астения и постуральная гипотензия или даже выраженная острая надпочечниковая недостаточность. Данный синдром редко встречается в первые 10 лет жизни, но может проявить себя на втором десятилетии или позже.
Циркулирующие АТл к желудочным париетальным клеткам и к внутреннему фактору у пациентов с СД-1 встречаются в 2-3 раза чаще, чем в популяции. Присутствие АТл к желудочным париетальным клеткам связано с атрофическим гастритом, а АТл к внутреннему фактору — с нарушением всасывания витамина В12. Мегалобластическая анемия у детей с СД-1 встречается редко.