а) Синдром Барттера. Синдром Барттера — это группа заболеваний, характеризующихся гипокалиемическим гипохлоремическим метаболическим алкалозом с гиперкальциурией и солевым истощением.
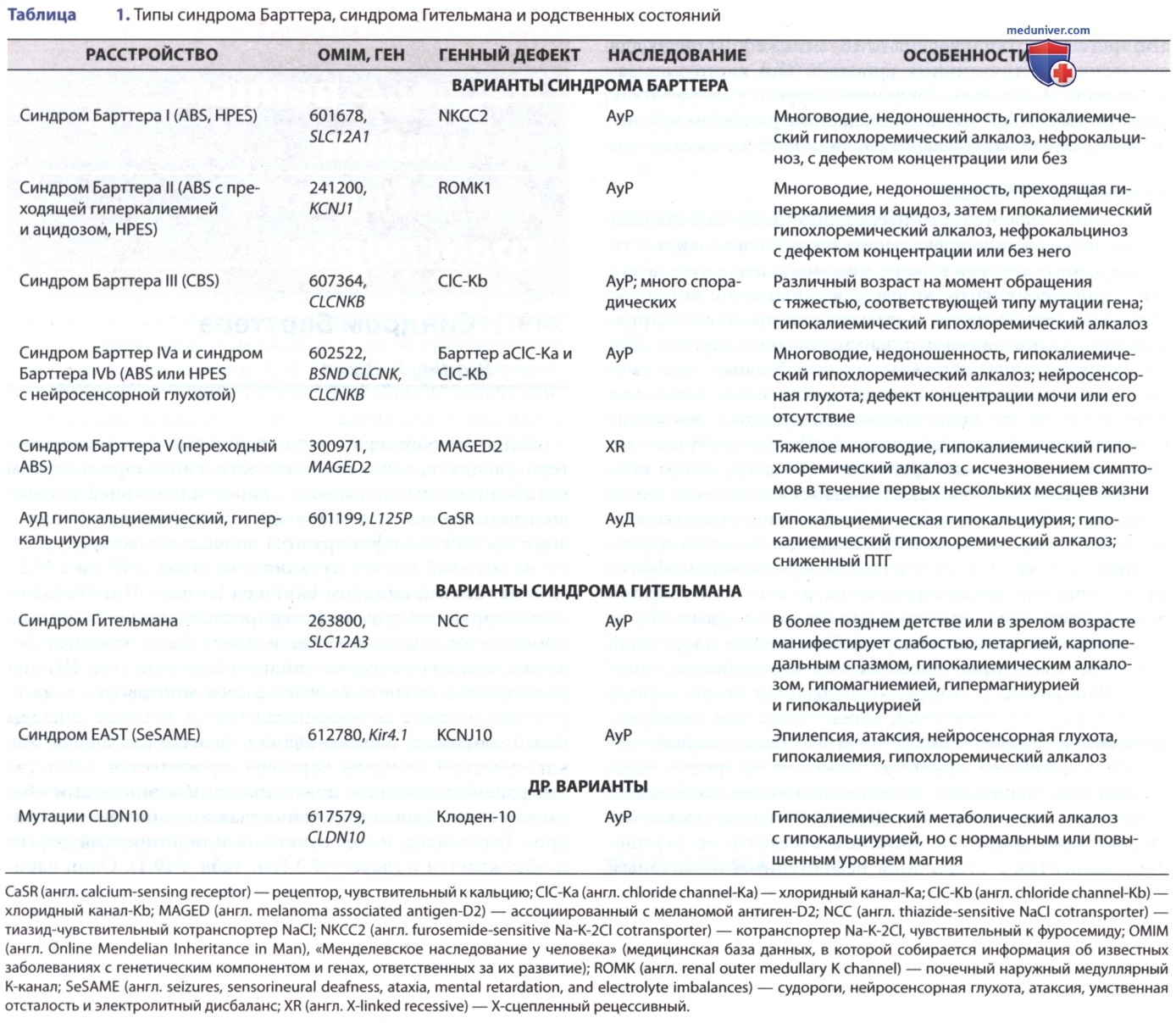
Эти нарушения в настоящее время классифицируются по анатомическому участку, на который влияет мутация гена (табл. 1 и 2). Антенатальный синдром Барттера (типы I, II и IV; также называемый синдромом гиперпростагландина Е) обычно проявляется в младенчестве и имеет более тяжелый фенотип, чем классический синдром Барттера (тип III); перинатальное начало включает в себя многоводие у матери, неонатальное солевое истощение и тяжелые эпизоды повторяющегося обезвоживания.
Более мягкий фенотип, классический синдром Барттера, проявляется в детстве задержкой развития и повторяющимися эпизодами обезвоживания. Фенотипически связанное заболевание, синдром Гительмана, имеет отчетливый генетический дефект (см. табл. 1). Один вариант антенатального синдрома Барттера связан с нейросенсорной глухотой (тип IV). Фенотипы, подобные Барттеру, были отмечены при др. заболеваниях, таких как синдром Кернса-Сейра.
1. Патогенез. Биохимические особенности синдрома Барттера, гипокалиемического гипохлоремического метаболического алкалоза с гиперкальциурией, напоминают те, которые наблюдаются при постоянном применении петлевых диуретиков, и отражают нарушение транспорта натрия, хлоридов и калия в восходящей петле Генле. Потеря натрия и хлоридов с результирующим обезвоживанием стимулирует ось РААС.
Альдостерон способствует захвату натрия и секреции калия, усугубляя гипокалиемию. Он также стимулирует секрецию ионов водорода дистально, ухудшая метаболический алкалоз. Гипокалиемия стимулирует синтез Pg, который дополнительно активирует ось РААС. Синдром Барттера ассоциирован, по крайней мере, с пятью различными генетическими дефектами транспортеров петли Генле (см. табл. 1). Каждый из них способствует переносу натрия и хлоридов.
Мутации в генах, которые кодируют транспортер натрия / калия / хлоридов (NKCC2, локализация действия фуросемида), просветный калиевый канал (ROMK), комбинированный хлоридный канал (CLC-Ka, CLC-Kb) или субъединицу каналов хлорида (барттин) вызывают неонатальный синдром Барттера. Изолированные дефекты генов, которые продуцируют специфический базолатеральный хлоридный канал (СlС-Kb), вызывают классический синдром Барттера.
2. Клинические проявления. В анамнезе м.б. выявлено многоводие у матери с недоношенностью или без. При медицинском осмотре могут присутствовать дисморфические черты лица, включая треугольное лицо, выступающие уши, большие глаза с косоглазием и отвисший рот. Родство предполагает наличие АуР-заболевания. У детей старшего возраста в анамнезе м.б. повторяющиеся эпизоды полиурии с обезвоживанием, задержкой роста, слабостью, головокружением и хроническими запорами, а также мышечными судорогами, вторичными по отношению к хронической гипокалиемии.
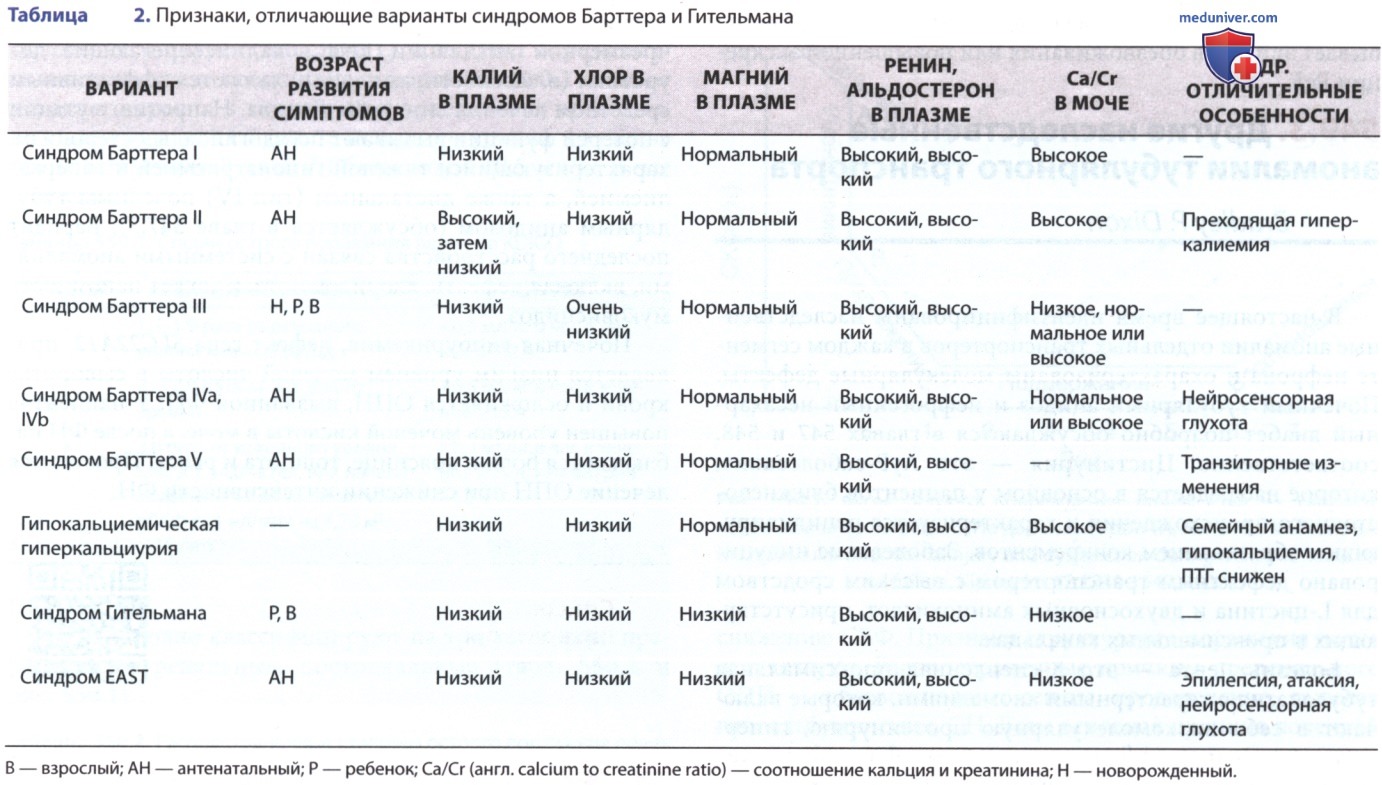
АД обычно в норме, хотя у пациентов с антенатальной формой м.б. сильное солевое истощение, приводящее к обезвоживанию и гипотонии. Химический анализ сыворотки выявляет классические биохимические аномалии гипокалиемического гипохлоремического метаболического алкалоза. Почечная функция обычно нормальная. Уровни кальция в моче часто повышены, как и уровни калия и натрия. Уровни ренина, альдостерона и PgE в сыворотке часто значительно повышены, особенно при более тяжелой антенатальной форме. Нефрокальциноз, возникающий вследствие гиперкальциурии, можно диганостировать по данным УЗИ (типы I и II).
3. Диагностика. Диагноз обычно основывается на клинической картине и лабораторных данных. У новорожденного или ребенка грудного возраста в клинической картине преобладают тяжелая гипокалиемия, обычно <2,5 ммоль/л, с метаболическим алкалозом. Гиперкальциурия типична; гипомагниемия наблюдается у меньшинства пациентов, но чаще встречается при синдроме Гительмана.
Поскольку признаки синдрома Барттера напоминают хроническое применение петлевых диуретиков, злоупотребление диуретиками следует учитывать при ДД даже у маленьких детей. Хроническая рвота и муковисцидоз также могут давать схожую клиническую картину, но ее можно отличить посредством определения уровня хлорида в моче, который повышен при синдроме Барттера и низок у пациентов с хронической рвотой и муковисцидозом. В почках отмечается гиперплазия юкстагломерулярного аппарата, хотя биопсия для диагностики этого состояния проводится редко.
4. Лечение и прогноз. Лечение синдрома Барттера направлено на предотвращение обезвоживания, поддержание статуса питания и коррекцию гипокалиемии. Требуется добавление калия, обычно в форме калия хлорида для коррекции сопутствующего истощения по показателю содержания хлоридов, и часто в очень высоких дозах. Даже при соответствующей терапии уровень калия в сыворотке крови м.б. низким, особенно у пациентов с неонатальной формой. Младенцы и дети раннего возраста нуждаются в диете с высоким содержанием натрия и, иногда, в БАД с натрием.
Индометацин, ингибитор Pg, также м.б. эффективным. Если присутствует гипомагниемия, необходимы БАД с магнием. При пристальном внимании к электролитному балансу, волюмическому статусу и росту долгосрочный прогноз в целом благоприятный. У меньшинства пациентов хроническая гипокалиемия, нефрокальциноз и хроническая терапия индометацином могут привести к хроническому интерстициальному нефриту и ХПН.
б) Синдром Гительмана. Синдром Гительмана (часто называемый «вариантом синдрома Барттера») — редкая АуР-причина гипокалиемического гипохлоремического метаболического алкалоза с отчетливыми признаками гипокальциуриии и гипомагниемии. Первые симптомы синдрома Гительмана обычно появляются в позднем детском или раннем взрослом возрасте (см. табл. 1 и 2).
1. Патогенез. Биохимические особенности синдрома Гительмана напоминают таковые при хроническом применении тиазидных диуретиков. Тиазиды действуют на котранспортер хлорида натрия NCCT, присутствующий в дистальных извитых канальцах. Посредством анализа сцепления и мутационных исследований у пациентов с синдромом Гительмана были продемонстрированы дефекты в гене, кодирующем NCCT.
2. Клинические проявления. Пациенты с синдромом Гительмана обычно выявляются в более старшем возрасте, чем пациенты с синдромом Барттера, и могут иметь симптомы, похожие на симптомы у детей старшего возраста с синдромом Барттера. Пациенты часто имеют в анамнезе повторяющиеся мышечные судороги и спазмы, предположительно вызванные низким уровнем магния в сыворотке крови, никтурией, полиурией и периодической гипотонией. Обычно у них нет в анамнезе повторяющихся эпизодов обезвоживания. Биохимические нарушения включают в себя гипокалиемию, метаболический алкалоз и гипомагниемию.
Уровень кальция в моче обычно очень низкий (в отличие от повышенного уровня кальция в моче, часто наблюдаемого при синдроме Барттера), а уровень магния в моче повышен. Уровни ренина и альдостерона обычно в норме, а секреция PgE не повышена. Нарушение роста менее выражено при синдроме Гительмана, чем при синдроме Барттера.
3. Диагностика. Диагноз синдрома Гительмана предполагается у подростков или взрослых с гипокалиемическим гипохлоремическим метаболическим алкалозом, гипомагниемией и гипокальциурией.
4. Лечение. Терапия направлена на коррекцию гипокалиемии и гипомагниемии с помощью дополнительных ЛП калия и магния. БАД с натрием или лечение ингибиторами Pg, как правило, не требуется, поскольку у пациентов обычно не бывает эпизодов обезвоживания или повышенной экскреции PgE.
в) Другие наследственные аномалии тубулярного транспорта. В настоящее время идентифицированы наследственные аномалии отдельных транспортеров в каждом сегменте нефрона и охарактеризованы молекулярные дефекты. Почечный тубулярный ацидоз и нефрогенный несахарный диабет подробно обсуждаются в отдельных статьях на сайте - просим Вас пользоваться формой поиска по сайту выше. Цистинурия — это АуР-заболевание, которое наблюдается в основном у пациентов ближневосточного происхождения и характеризуется рецидивирующим образованием конкрементов.
Заболевание индуцировано дефектным транспортером с высоким сродством для L-цистина и двухосновных аминокислот, присутствующих в проксимальных канальцах.
Болезнь Дента — это Х-сцепленная проксимальная тубулопатия с характерными аномалиями, которые включают в себя низкомолекулярную протеинурию, гиперкальциурию и др. признаки синдрома Фанкони, такие как глюкозурия, аминоацидурия и фосфатурия. Хотя у некоторых пациентов развивается нефрокальциноз, нефролитиаз, прогрессирующая ХПН и гипофосфатемический рахит, пациенты с болезнью Дента обычно не имеют проксимального почечного канальцевого ацидоза или внепочечных проявлений.
Мутации потери функции гена CLCN5, который кодирует почечный антипортер хлоридов/водорода (ClC-5), имеется у 50-60% пациентов с болезнью Дента. Генетйческая гетерогенность болезни Дента у некоторых пациентов с мутациями в гене OCRL1 (ответственного за синдром Лоу) также соответствует критериям болезни Дента (15% пациентов): болезнь Дента 2.
Заболевание включает Х-сцепленный рецессивный нефролитиаз с ХПН, Х-сцепленный рецессивный гипофосфатемический рахит и идиопатическую низкомолекулярную протеинурию, наблюдаемую у японских детей.
Мутации во внеклеточном базолатеральном кальциевом рецепторе, обычно присутствующем в петле Генле, могут вызывать доминантную картину, подобную синдрому Барттера (также известную как синдром Барттера типа V). Преобладающими симптомами у этих пациентов являются гипокальциемия и подавление функции ПТГ, что отличает их от пациентов с синдромом Барттера.
В дистальном извитом канальце мутации с усилением функции в WNK1 и мутации с потерей функции в WNK4, обе сериновые треонинкиназы, приводят к чрезмерной NCCT-опосредованной реабсорбции соли с клинической картиной псевдогипоальдостеронизма типа 2 (семейная гиперкалиемическая гипертензия, или синдром Гордона), включая увеличение ОЦК с АГ, гиперкалиемией, гиперхлоремическим метаболическим ацидозом и гиперкальциурией. Вследствие чрезмерной активации тиазид-чувствительного NCCT это заболевание можно эффективно лечить с помощью тиазидных диуретиков.
Мутации с усилением функции гена, кодирующего эпителиальный натриевый канал (ENaC) в собирательных трубочках, вызывают наследственную форму АГ, синдром Лиддла. У пациентов с этим заболеванием наблюдается поглощение натрия в собирательных трубочках, гипокалиемия и подавление секреции альдостерона. Вследствие чрезмерной активации ENaC, калийсберегающие диуретики (в частности, амилорид) являются эффективным средством лечения синдрома Лиддла.
Напротив, мутации с потерей функции вызывают псевдогипоальдостеронизм, характеризующийся тяжелой гипонатриемией и гиперкалиемией, а также дистальным (тип IV) почечным тубулярным ацидозом. Вариант последнего расстройства связан с системными аномалиями, включая дефекты хлоридов пота, и может напоминать муковисцидоз.
Почечная гипоурикемия, дефект гена SLC22A12, проявляется низким уровнем мочевой кислоты в сыворотке крови и осложняется ОПН, вызванной ФН. У пациентов повышен уровень мочевой кислоты в моче, а после ФН наблюдаются боли в пояснице, тошнота и рвота. Проводится лечение ОПН при снижении интенсивности ФН.