Почечный тубулярный ацидоз (ПТА) — заболевание, характеризующееся неанионным (гиперхлоремическим) метаболическим ацидозом при нормальной или почти нормальной СКФ. Выделяют 4 основных типа: проксимальный (тип II) ПТА, классический дистальный (тип I) ПТА, гиперкалиемический (тип IV) ПТА и комбинированный проксимальный и дистальный (тип III). Проксимальный ПТА возникает в результате нарушения реабсорбции бикарбонатов, а дистальный ПТА — в результате неспособности секретировать кислоту. Любой из этих дефектов может передаваться по наследству и быть врожденным или приобретенным, что чаще встречается в клинической практике.
а) Нормальное подкисление мочи. Почки вносят вклад в КЩС за счет реабсорбции фильтрованного бикарбоната (НСО3-) и выведения иона водорода (Н+), образующегося каждый день. Секреция Н+ кл. канальцев в просвет играет ключевую роль в реабсорбции НСО3- и образовании титруемой кислоты (Н+ связываются с буферами, такими как НРО42-) и ионами аммония (NH4+).
Поскольку потеря отфильтрованного НСО3- эквивалентна добавлению Н+ в организм, весь отфильтрованный НСО3- должен абсорбироваться до того как полученные с пищей Н+ м.б. экскретированы. Примерно 90% отфильтрованного НСО3- абсорбируется в проксимальных канальцах, а оставшиеся 10% — в дистальных сегментах, в основном в толстом восходящем колене петли Генле и наружном медуллярном собирательном канальце (см. рис. 1).
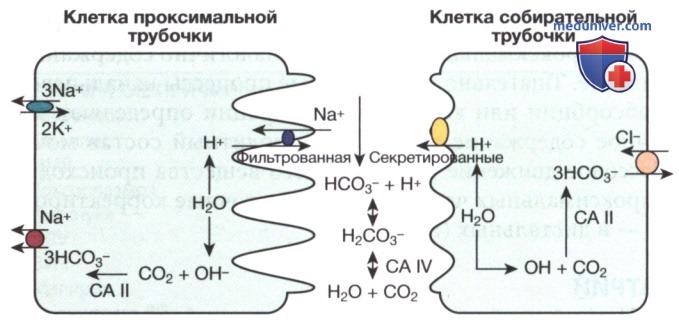
Рисунок 1. Основные события, происходящие в кл. проксимальных и собирательных трубочек с целью поддержания кислотно-щелочного состояния. В проксимальной трубочке Н+, отделяясь от воды, секретируется в просвет посредством натриево-водородного обменника; НСО3- формируется вследствие объединения ОН" (из молекулы воды) и СО2 в присутствии карбоангидразы II, и возвращается в системный кровоток посредством натриево-бикарбонатного котранспортера. Аналогично, в собирательной трубочке Н+ секретируется в просвет благодаря Н+АТФазе; и НСО3- возвращается в системный кровоток посредством бикарбонатно-хлоридного обменника. Ионы водорода секретируются в просвет в комбинации с фильтрованным НСО3- с целью формирования Н2СО3, а затем воды и СО2в присутствии карбоангидразы IV, которые могут пассивно реабсорбироваться
В проксимальном канальце и толстом восходящем колене петли Генле Н+ из воды секретируется натриево-водородным обменником на просветной мембране. Ионы водорода соединяется с фильтрованным НСО3-, что приводит к образованию угольной кислоты (Н2СО3), которая расщепляется на воду и углекислый газ (СО2) в присутствии карбоангидразы IV. Углекислый газ свободно рассеивается обратно в кл., соединяется с анионами гидроксида (ОН-) из воды с образованием НСО3- в присутствии карбоангидразы II и возвращается в системный кровоток посредством натриево-бикарбонатного котранспортера, расположенного на базолатеральной мембране кл. В собирательном канальце Н+ секретируется в просвет с помощью Н+АТФазы, а НСО3- возвращается в системный кровоток с помощью бикарбонатно-хлоридного обменника, расположенного на базолатеральной мембране.
Ионы водорода, избыточно секретируемые вследствие фильтрации НСО3- проксимально и дистально, выводятся с мочой либо в виде титруемой кислоты (Н2РО4-), либо в виде NH4+.
1. Патогенез. Проксимальный ПТА может передаваться по наследству и присутствовать с рождения или возникать как временное явление в младенчестве. Хотя и редко, но м.б. первичным и изолированным. Проксимальный ПТА обычно возникает как компонент глобальной проксимальной канальцевой дисфункции или синдрома Фанкони, который характеризуется низкомолекулярной протеинурией, глюкозурией, фосфатурией, аминоацидурией и проксимальным ПТА. В табл. 1 указаны причины проксимального ПТА и синдрома Фанкони. Многие из этих причин являются наследственными заболеваниями.
В дополнение к цистинозу и синдрому Лоу АуР и АуД проксимальный ПТА рассматриваются далее в этом разделе. Др. наследственные формы синдрома Фанкони включают в себя галактоземию, наследственную непереносимость фруктозы, тирозинемию и болезнь Вильсона.
Болезнь Дента, или Х-сцепленный нефролитиаз, обсуждается в отдельной статье на сайте - просим Вас пользоваться формой поиска по сайту выше. У детей важной формой вторичного синдрома Фанкони является воздействие ЛС, таких как ифосфамид, который является компонентом многих схем лечения опухоли Вильмса и др. ЗНО.
- Аутосомно-рецессивное заболевание. Изолированный аутосомно-рецессивный проксимальный ПТА индуцирован мутациями в гене, кодирующем котранспортер бикарбоната натрия NBC1. Он проявляется глазными аномалиями (ленточная кератопатия, катаракта и глаукома, часто приводящими к слепоте), низким ростом, дефектами эмали зубов, умственными нарушениями и иногда кальцификацией базальных ганглиев наряду с проксимальным ПТА. АуД-тип наследования был идентифицирован в единственной родословной с девятью членами с гиперхлоремическим метаболическим ацидозом, нормальной способностью подкислять мочу, нормальной функцией почек и задержкой роста.
- Цистиноз. Цистиноз — системное заболевание, вызванное дефектом метаболизма цистеина, который приводит к накоплению кристаллов цистина (окисленная форма цистеина, в которой две молекулы цистеина соединены своими сульфгидрильными группами посредством дисульфидной связи) в большинстве органов, особенно в почках, печени, глазах и ГМ. Заболеваемость составляет 1:100 000-1: 200 000. В определенных группах населения, напр., у французских канадцев, заболеваемость намного выше. Описаны, по крайней мере, три клинических картины.
У маленьких детей наиболее тяжелая форма заболевания (инфантильный или нефропатический цистиноз) проявляется на первом/втором годах жизни тяжелой дисфункцией канальцев и задержкой роста.
Если заболевание не лечить, к концу первого десятилетия у детей развивается терминальная стадия ХПН. Более легкая форма заболевания проявляется у подростков и характеризуется менее тяжелыми аномалиями канальцев и более медленным прогрессированием до ХПН. Также существует доброкачественная взрослая форма с поражением глаз, но без поражения почек.
Цистиноз индуцирован мутациями в гене CTNS, который кодирует белок цистинозин. Считается, что цистинозин является Н+-управляемым лизосомальным переносчиком цистина. Фенотипические и генотипические исследования демонстрируют, что пациенты с тяжелым нефропатическим цистинозом несут мутации, которые приводят к полной потере функции цистинозина. У пациентов с более легким течением заболевания наблюдаются мутации, которые приводят к экспрессии частично функционального белка. Пациенты с нефропатическим цистинозом имеют клинические проявления, отражающие их выраженную тубулярную дисфункцию и синдром Фанкони, включая полиурию и полидипсию, задержку роста и рахит. Часто бывает жар, вызванный обезвоживанием или уменьшением потоотделения.
Пациенты обычно светлокожие и светловолосые из-за уменьшения пигментации. Глазные проявления включают в себя светобоязнь, ретинопатию и снижение остроты зрения. У пациентов также может развиться гипотиреоз, гепатоспленомегалия и задержка полового созревания. При прогрессирующем тубулоинтерстициальном фиброзе почечная недостаточность инвариантна.
Диагноз цистиноза предполагает обнаружение кристаллов цистина в роговице и подтверждается определением повышенного содержания цистина в лейкоцитах. Для семей из группы риска доступно дородовое тестирование.
Лечение цистиноза направлено на коррекцию метаболических нарушений, связанных с синдромом Фанкони или ХПН. Кроме того, доступна специфическая терапия цистеамином, который превращает цистин в цистеин и гетеродимер цистеин-цистеамин. Это облегчает лизосомный транспорт и снижает уровень тканевого цистина. Цистеамин при пероральном приеме не достигает адекватных уровней в тканях глаза, поэтому требуется дополнительная терапия глазными каплями с цистеамином. Раннее начало приема ЛП может предотвратить или отсрочить ухудшение функции почек. Пациентам с задержкой роста, которая не улучшается с помощью цистеамина, может помочь лечение СТГ. Трансплантация почки — приемлемый вариант для пациентов с ХПН.
При увеличении выживаемости могут стать очевидными дополнительные осложнения, включая аномалии ЦНС, мышечную слабость, дисфагию и недостаточность ПЖЖ. Неясно, уменьшит ли длительная терапия цистеамином эти осложнения.
- Синдром Лоу. Синдром Лоу (окуло-церебро-ренальный синдром) — редкое Х-сцепленное заболевание, характеризующееся врожденной катарактой, умственной отсталостью и синдромом Фанкони. Заболевание вызвано мутациями в гене OCRL1, который кодирует белок фосфатидилинозитолполифосфат-5-фосфатазу. Считается, что аномалии, наблюдаемые при синдроме Лоу, вызваны аномальным транспортом пузырьков в аппарате Гольджи. В почках наблюдаются неспецифические тубулоинтерстициальные изменения. Также наблюдаются утолщение гломерулярной базальной мембраны и изменения митохондрий проксимальных канальцев.
Пациенты с синдромом Лоу в младенчестве обычно имеют катаракту, прогрессирующую задержку роста, гипотонию и синдром Фанкони. Часто встречается значительная низкомолекулярная протеинурия. У пациентов развиваются слепота и ХПН. Также наблюдаются характерные поведенческие отклонения, включая истерики, упрямство, стереотипию (повторяющееся поведение) и навязчивые идеи. Специфической терапии ХПН или неврологического дефицита не существует. Обычно требуется экстракция катаракты.
2. Клинические проявления проксимального почечного тубулярного ацидоза и синдрома Фанкони. У пациентов с изолированным, спорадическим или наследственным проксимальным ПТА наблюдается задержка роста на 1-м году жизни. Дополнительные симптомы могут включать в себя полиурию, обезвоживание (вследствие потери натрия), анорексию, рвоту, запор и гипотонию. Пациенты с первичным синдромом Фанкони имеют дополнительные симптомы, вторичные по отношению к фосфатному истощению, такие как рахит. У пациентов с системными заболеваниями появляются дополнительные признаки и симптомы, характерные для их основного заболевания. ОАМ у пациентов с изолированным проксимальным ПТА обычно без особенностей.
Кислотность мочи низкая (<5,5), т.к. у этих пациентов не нарушены механизмы дистального подкисления. Исследования мочи у пациентов с синдромом Фанкони демонстрируют разную степень фосфатурии, аминоацидурии, глюкозурии, урикозурии и повышенное содержание натрия или калия. В зависимости от характера основного заболевания могут присутствовать лабораторные доказательства ХПН, включая увеличение креатинина сыворотки.
в) Дистальный (тип I) почечный тубулярный ацидоз:
1. Патогенез. Дистальный ПТА м.б. спорадическим или наследственным. Он также может возникать как осложнение наследственных или приобретенных заболеваний дистальных канальцев. Первичные или вторичные причины дистального ПТА м.б. результатом повреждения или нарушения функционирования одного или нескольких транспортеров или белков, участвующих в процессе подкисления, включая Н+АТФазу, бикарбонатно-хлоридный обменник или компоненты РААС. Вследствие нарушения экскреции ионов водорода pH мочи не м.б. снижена до 5,5, несмотря на наличие тяжелого метаболического ацидоза. Потеря бикарбоната натрия дистально из-за отсутствия Н+ для связывания в просвете канальцев (см. рис. 1) приводит к увеличению абсорбции хлоридов и гиперхлоремии.
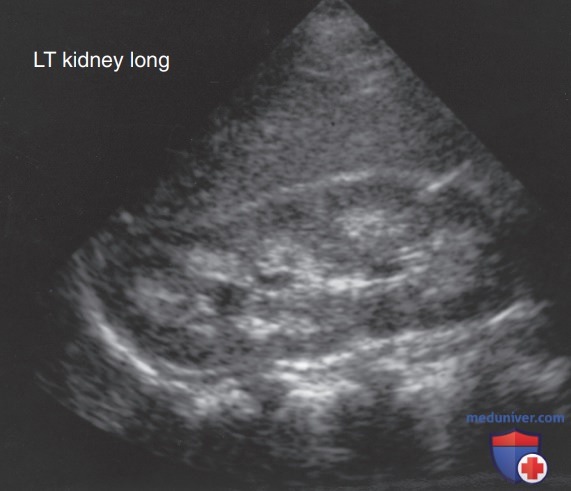
Рисунок 2. Ультразвуковое исследование ребенка с дистальным почечным тубулярным ацидозом, при котором визуализируется медуллярный нефрокальциноз
Неспособность секретировать Н+ компенсируется повышенной секрецией калия дистально, что приводит к гипокалиемии. Обычно присутствует гиперкальциурия, которая может привести к нефрокальцинозу или нефролитиазу. Хронический метаболический ацидоз также ухудшает выведение цитрата с мочой. Гипоцитратурия дополнительно увеличивает риск отложения кальция в канальцах. Заболевание костей является распространенным явлением, возникающим в результате мобилизации органических компонентов из кости, которые служат буфером для хронического ацидоза.
2. Клинические проявления. Дистальный ПТА имеет общие черты с таковыми проксимального ПТА, включая метаболический ацидоз без дисбаланса анионов и нарушение роста. Отличительные признаки дистального ПТА включают в себя нефрокальциноз и гиперкальциурию. Фосфаты и массивная потеря НСО3-, характерная для проксимального ПТА, обычно отсутствуют. В табл. 2 перечислены причины первичного и вторичного дистального ПТА. Хотя наследственные формы встречаются редко, были идентифицированы три специфические наследуемые формы дистального ПТА, включая АуР-форму, связанную с нейросенсорной глухотой.
Медуллярная губчатая почка — относительно редкое спорадическое заболевание у детей, у взрослых встречается чаще. Оно характеризуется кистозным расширением концевых частей собирательных трубочек при их входе в почечные пирамиды. При УЗИ у пациентов часто наблюдается медуллярный нефрокальциноз. Хотя пациенты с этим заболеванием обычно поддерживают нормальную функцию почек в зрелом возрасте, осложнения включают в себя нефролитиаз, пиелонефрит, гипостенурию (неспособность концентрировать мочу) и дистальный ПТА. Сообщалось о связи медуллярной губчатой почки с синдромом Беквита-Видемана или гемигипертрофией.
1. Патогенез. ПТА IV типа возникает в результате нарушения выработки альдостерона (гипоальдостеронизм) или нарушения реакции почек на альдостерон (псевдогипоальдостеронизм). Ацидоз возникает вследствие того, что альдостерон напрямую влияет на Н+АТФазу, ответственную за секрецию Н+. Кроме того, альдостерон является мощным стимулятором секреции калия в собирательных трубочках; следовательно, его недостаток приводит к гиперкалиемии. Это дополнительно влияет на КЩС, ингибируя аммониогенез и, таким образом, выведение Н+. Дефицит альдостерона обычно возникает в результате нарушений надпочечников, таких как болезнь Аддисона или некоторых форм врожденной гиперплазии надпочечников. У детей отсутствие реакции на альдостерон является более частой причиной ПТА типа IV.
Это может происходить временно, напр., при эпизоде острого пиелонефрита или острой непроходимости МВП, или хронически, особенно у младенцев и детей с обструктивной уропатией в анамнезе. У последних пациентов м.б. значительная гиперкалиемия даже в тех случаях, когда функция почек нормальна или нарушена незначительно. Выявлены редкие примеры наследственных форм ПТА типа IV (см. табл. 3).
2. Клинические проявления. Пациенты с ПТА IV типа могут иметь задержку роста в первые несколько лет жизни. Часто встречаются полиурия и обезвоживание (вследствие потери солей). В редких случаях у пациентов (особенно с псевдогипоальдостеронизмом 1 типа) развивается жизнеугрожающая гиперкалиемия. У пациентов с обструктивными уропатиями могут возникать симптомы острого пиелонефрита, такие как лихорадка, рвота и неприятный запах мочи. Лабораторные исследования выявляют гиперкалиемический метаболический ацидоз с нормальным анионным составом. Моча м.б. щелочной или кислой. Повышенный уровень натрия в моче при аномально низком уровне калия отражает отсутствие эффекта альдостерона.
3. Диагностика почечного тубулярного ацидоза. Первым шагом в обследовании пациента с подозрением на ПТА является подтверждение наличия метаболического ацидоза с нормальным анионным интервалом, выявление нарушений электролитного баланса, оценка функции почек и исключение др. причин потери НСО3-, таких как диарея (табл. 4).
Метаболический ацидоз, связанный с диарейным обезвоживанием, чрезвычайно распространен, состояние обычно улучшается после коррекции ОЦК. Пациенты с затяжной диареей могут истощать запасы НСО3- в организме и иметь стойкий ацидоз, несмотря на очевидное восстановление ОЦК. В случаях, когда у пациента в недавнем анамнезе была тяжелая диарея, полное обследование на ПТА следует отложить на несколько дней, чтобы дать достаточно времени для восстановления запасов НСО3- в организме. Если в этих условиях ацидоз сохраняется более нескольких дней, необходимы дополнительные исследования.
Электролиты сыворотки, AMK, кальций, фосфор, креатинин и pH должны быть получены путем венепункции. Травматический забор крови (напр., образцы пяточной пункции), небольшие объемы крови в пробирках для взятия образцов у взрослых, или длительное время транспортировки образцов при комнатной температуре могут привести к ложно низкому уровню НСО3-, часто в сочетании с повышенным содержанием калия в сыворотке. Истинный гиперкалиемический ацидоз согласуется с ПТА типа IV, тогда как обнаружение нормального или низкого калия предполагает тип I или II. Анионный интрвал крови следует рассчитывать по формуле: [Na+] - [Cl- + НСО3-], где Na+ — уровень натрия в плазме крови, Cl — уровень хлоридов в плазме крови, НСО3- — уровень бикарбоната в плазме крови. Значения <12 демонстрируют отсутствие анионного интервала.
Значения >20 указывают на наличие анионного интервала. Если наличие анионного интервала верифицировано, следует исключить др. причины (лактатацидоз, диабетический кетоацидоз, врожденные нарушения метаболизма, интоксикация). Если отмечается тахипноэ, оценка газового состава артериальной крови м.б. подходящей для исключения возможного смешанного кислотно-основного нарушения, в первую очередь затрагивающего респираторные и метаболические компоненты. Очень важен подробный анамнез с особым вниманием к росту и развитию; недавним или повторяющимся заболеваниям, сопровождающимся диареей; семейному анамнезу: умственной отсталости, задержке развития, терминальной стадии ХПН, детской смертности или наличию выкидышей. При физикальном обследовании необходимо определить параметры роста и волюмический статус, а также наличие любых дисморфических особенностей, указывающих на основной синдром.
После подтверждения наличия метаболического ацидоза без анионного интервала, pH мочи может помочь отличить дистальную причину от проксимальной. Кислотность мочи <5,5 при наличии ацидоза указывает на проксимальный ПТА, тогда как у пациентов с дистальным ПТА обычно pH мочи >6,0. Анионный интервал мочи [(Na++K+)-Cl-, где Na+, содержание натрия в моче; К+, содержание калия в моче; Cl-, содержание хлоридов в моче] м.б. рассчитан для подтверждения диагноза дистального ПТА. Положительный интервал указывает на недостаточность аммониогенеза и, следовательно, на возможность дистального ПТА.
Отрицательный соответствует истощению НСО3- проксимальных канальцев (или истощению НСО3- ЖКТ). Также следует провести ОАМ, чтобы определить наличие глюкозурии, протеинурии или гематурии, предполагая более глобальное повреждение или дисфункцию канальцев. Спонтанные или суточные измерения кальция и креатинина в моче позволят выявить гиперкальциурию. УЗИ почек следует выполнять для выявления основных структурных аномалий, таких как обструктивные уропатии, а также для определения наличия нефрокальциноза (см. рис. 2).
4. Лечение и прогноз. Основа терапии при всех формах ПТА — заместительная терапия НСО3-. Пациентам с проксимальным ПТА часто требуется большое количество НСО3-, до 20 моль-эквивалент/кг в сутки, в форме р-ра натрия гидрокарбоната («Натрия бикарбоната») или натрия цитрата. Базовое требование для дистального ПТА обычно находится в диапазоне 2-4 моль-эквивалент/кг в сутки, хотя индивидуальные потребности пациентов могут варьировать. Пациентам с синдромом Фанкони обычно требуется дополнительный прием фосфатов. Пациентов с дистальным ПТА следует наблюдать на предмет развития гиперкальциурии. При наличии симптоматической гиперкальциурии (повторяющиеся эпизоды макрогематурии), нефрокальциноза или нефролитиаза могут потребоваться тиазидные диуретики для снижения выведения кальция с мочой.
Пациентам с ПТА IV типа может потребоваться длительное лечение гиперкалиемии натрий-калиевой обменной смолой (напр., полистирол сульфонатом натрия).
Прогноз при ПТА во многом зависит от характера имеющегося основного заболевания. Пациенты с леченым изолированным проксимальным или дистальным ПТА обычно демонстрируют улучшение роста при условии, что уровни НСО3- в сыворотке могут поддерживаться в нормальном диапазоне. Пациенты с системным заболеванием и синдромом Фанкони могут иметь постоянные проблемы с задержкой роста, рахитом, а также признаками и симптомами, связанными с их основным заболеванием.
д) Рахит, ассоциированный с почечным тубулярным ацидозом. Рахит может присутствовать при первичном ПТА, особенно при проксимальном ПТА, вследствие гипофосфатемии и фосфатурии, вызванных генерализованной дисфункцией проксимальных канальцев. Деминерализация костей без явного рахита обычно выявляется при дистальном (тип I) ПТА. Это метаболическое заболевание может характеризоваться болью в костях, задержкой роста, остеопенией и иногда патологическими переломами.
Деминерализация костей при дистальном ПТА, вероятно, связана с лизисом кости, потому что карбонат кальция в кости служит буфером против метаболического ацидоза вследствие задержки выведения Н+.
Введение достаточного количества НСО3-, чтобы купировать ацидоз, уменьшает остепороз и гиперкальциурию, которая часто встречается при дистальном ПТА. При проксимальном ПТА возможна терапия как бикарбонатными, так и пероральными фосфатными БАД для лечения рахита. Могут использоваться дозы фосфатов, аналогичные тем, которые применяются при лечении семейной гипофосфатемии или синдроме Фанкони. Витамин D необходим для компенсации вторичного гиперпаратиреоза, который усложняет пероральную фосфатную терапию. После терапии темпы роста у пациентов с ПТА типа II (проксимальный) выше, чем у пациентов с первичным синдромом Фанкони.
Видео респираторный и метаболический ацидозы в анализах