Атаксия, гиперурикемия и почечная недостаточность с глухотой
Синдром, характеризующийся гиперурикемией, почечной недостаточностью, атаксией и глухотой, был описан у 5 больных из трех поколений одной семьи (Rosenberg, Bergstrom, Troost, Bartholomew). У б других членов семьи наблюдались различные компоненты синдрома.
Клинические данные. Нервная система. У 6 больных во втором и третьем десятилетии жизни была впервые отмечена атаксическая походка. В дальнейшем нарушения координации медленно прогрессировали и охватывали как ноги, так и руки. Частым симптомом была также дизартрия. У одного больного при взгляде в стороны наблюдался горизонтальный нистагм. У 5 из 6 больных с атаксией отмечались явные атрофии или слабость проксимальной мускулатуры конечностей. Сухожильные рефлексы были повышены.
У 2 больных наблюдались легкие нарушения вибрационной чувствительности в дистальных отделах конечностей, в остальном чувствительность была нормальной.
Сердечно-сосудистая система. На ЭКГ у пробанда обнаружено увеличение предсердия, отклонение оси сердца влево и синдром Вольффа — Паркинсона — Уайта (Wolff, Parkinson, White). В 22-летнем возрасте у больного развилась застойная сердечная недостаточность и появился ритм галопа. Это состояние было диагностировано как кардиомиопатия. У 43-летней матери больного обнаружена гипертрофия левого желудочка.
Орган слуха. Дефект слуха был впервые замечен во втором или третьем десятилетии жизни. У младшего больного обнаружена только высокочастотная нейросенсорная глухота, в то же время у старшего больного отмечалась резко выраженная пейросенсорпая глухота с несомненным прогрессированием.
Различение речи значительно варьировало. Детальное аудиометрическое исследование, произведенное одному больному, позволило предположить кохлеарную локализацию глухоты.
Вестибулярная система. Всем больным с атаксией была произведена электропистагмографпя. У двоих обнаружено патологическое маятникообразное ускорение. В ответ па калорическую стимуляцию у одного из них появились патологические глазодвигательные реакции, а у другого — резкая тошнота и рвота.
Лабораторные данные. Произведенное всем больным энзиматическое исследование выявило интактность эритроцитарной гипоксантин-гуанин-фосфорибозилтрансферазы (ТГФРТ) и аденин-фосфорибозилтрансферазы (АФРТ).
У 9 членов семьи была обнаружена гпперурикемия. Семи из них были произведены функциональные почечные пробы, у пяти пробы клиренса РАН, креатинина и инсулина были в пределах нормы или несколько снижены. Согласно данным клиренса креатинина, у 4 больных с гиперуриксмией функция почек была нормальной, что позволило заподозрить, что гиперурикемия не является вторичной, развившейся вследствие почечной недостаточности.
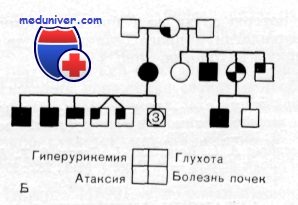
Наследственность. Больные были обнаружены в каждом из трех поколений семьи. Экспрессивность синдрома варьировала. У некоторых больных отмечалась только гиперурикемия, у других — глухота и атаксия. Очевидно гиперурикемия, атаксия и глухота являются разными проявлениями одного и того же плейотропного гена. Синдром наследуется по аутосомно-доминантиому типу.
Диагноз. При синдроме Алыюрта не отмечается ни гиперурикемпи, ни атаксии. Существует несколько наследственных синдромов, включающих в качестве характерных симптомов атаксию и глухоту, но неизвестно другого синдрома, при котором атаксия и глухота сочетались бы с гиперурикемией или почечной недостаточностью. Обсуждаемый здесь синдром отличается от болезни Леш — Пихана тем, что при нем не наблюдается самоповреждений, умственной отсталости, хореоатетоза, а также отсутствия активности ГТФРТ.
Отличаются также типы наследования. Синдром, описанный Kef ley с соавт., характеризующийся гиперурикемией, почечнокаменной болезнью и легкой мозжечковой атаксией с частичной недостаточностью фермента гипоксантин-гуанин-фосфорибозилтрансферазы (ГТФРТ), отличается от рассматриваемого здесь синдрома тем, что у этих больных имеется глухота и нет недостаточности ГТФРТ.
Лечение. Необходимо проводить симптоматическое лечение разных дефектов. При глухоте могут оказаться полезными слуховые аппараты, при гиперурикемпи можно применять аллопуринол.
Против атаксии и мышечных атрофии мало что может оказаться эффективным.
Прогноз. Хотя проявления атаксии и глухоты варьируют, оба симптома медленно прогрессируют и становятся резко выраженными. Гиперурикемия может привести к подагре, как это было обнаружено у старших членов семьи.
Выводы. Характеристика этого синдрома включает: 1) аутосомно-доминантное наследование с варьирующей экспрессивностью; 2) гиперурикемию; 3) начинающуюся во втором десятилетии жизни и очень медленно прогрессирующую атаксию; 4) почечную недостаточность; 5) потерю слуха по нейросенсорному типу, начинающуюся во втором десятилетии и прогрессирующую до глубокой глухоты, и 6) вестибулярную патологию.