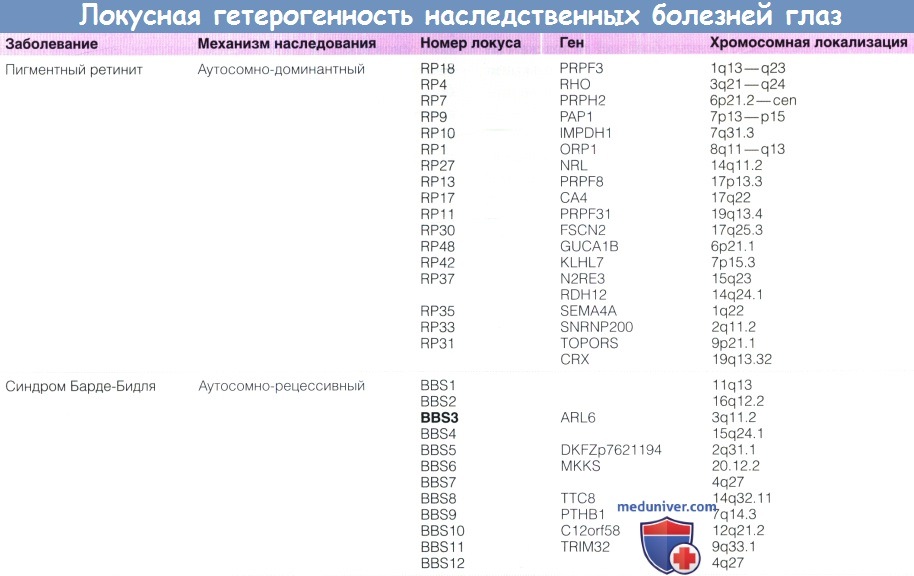
Генетическое консультирование при глазных болезнях
Пациенты и семьи обращаются за генетической консультацией, чтобы получить информацию о природе заболевания, риске развития заболевания или передаче его детям, о проблемах генетического обследования, рождения детей и лечения. Генетическое консультирование преследует цель помочь пациентам понять полученную информацию, избрать оптимальный способ действий и наилучшим образом адаптироваться к заболеванию.
Точная диагностика — главное условие эффективного генетического консультирования. Диагностика многих наследственных болезней глаз проводится по клиническим данным, для этого требуется участие специалистов-клиницистов и, часто, мультидисциплинарный подход, с проведением генетических, офтальмологических и электрофизиологических исследований.
Диагноз основывается на данных детального семейного анамнеза с построением родословного древа трех поколений, осмотра (часто нескольких членов семьи), а также на данных анамнеза болезни, включающих описание системных проявлений. Крайне важно проявлять настороженность в отношении глазных и экстраокулярных проявлений заболевания.
Генетическое консультирование при наследственных болезнях глаз может представлять собой особенно трудную задачу. Гетерогенность и перекрывание фенотипов затрудняет понимание диагноза пациентами. Многие наследственные заболевания сетчатки сопровождаются прогрессирующим ухудшением зрения и требуют предварительной адаптации к необходимости ухода. Коммуникативные потребности пациентов с нарушениями зрения требуют, чтобы информация поступала к ним в подходящем формате.
а) Генетические лабораторные тесты. Молекулярный анализ стал дешевле и доступнее, в настоящее время он применим в клинике. Клиницисту необходимо представлять себе его возможности. При моногенных наследственных болезнях глаз анализ, вероятно, будет заключаться в секвенировании генов. Анализы выполняются в качестве метода, дополняющего подробное клиническое обследование. Они проводятся с целью уточнения диагноза, например, при заболеваниях, характеризующихся крайней генетической гетерогенностью, неразличимых клинически.
В будущем генетическая диагностика может потребоваться для проведения геноспецифичного лечения (медикаментозного или генной терапии). Если оценка риска, например, при заболевании с доминантным наследованием, не вызывает затруднений, то для родственников пациента с доминантным фенотипом при сниженной пенетрантности (доминантная атрофия зрительного нерва и аутосомно-доминантная врожденная катаракта) или детей женщин из семьи, где мужчины страдают Х-сцепленным ретиношизисом она более сложная.
Молекулярный анализ выполняется на материале ДНК, выделенном из периферической крови или слюны одного больного пациента (пробанда) или более широкого круга родственников. После выявления патогенной мутации можно провести скрининг других членов семьи, в т.ч. неродившихся, на ее наличие.
б) Что такое мутация? Генетическая вариабельность является результатом процесса мутации ДНК. Описаны разнообразные механизмы мутаций при наследственных генетических и менделирующих болезнях человека. Большинство из них представляет собой феномен типа «все или ничего»: больные пациенты являются носителями патогенных генетических изменений («мутаций»), тогда как здоровые индивиды — нет. В таких случаях больные представители оной семьи являются носителями одинаковых генетических изменений, и эти изменения не меняются.
Однако существует небольшая группа заболеваний, к которой относится, например, миотоническая дистрофия, характеризующихся «динамическими» мутациями, при которых генетические изменения в разных поколениях одной семьи могут варьировать.
1. Хромосомные альтерации. Наиболее грубыми генетическими изменениями являются альтерации на уровне хромосом, а именно цитогенетически визуализируемые перестройки, такие как делеции, инверсии, дупликации и транслокации. Подобный «геномный дисбаланс» очень плохо переносится, и за все время проводимых исследований наблюдалась лишь незначительная часть всех возможных перестроек. Такие изменения включают в себя трисомии (например, трисомию 21 или синдром Дауна), а также бширные хромосомные делеции (например, хромосомную делецию 11р, вызывающую синдром WAGR, см. выше).
2. Субмикроскопические геномные перестройки. В настоящее время возможно сравнить тонкие различия в количестве копий ДНК между разными индивидами. «Субмикроскопические геномные перестройки» включают в себя как утрату генетического материала (микроделеции), так и увеличение его количества (микродупликации) и являются причинами наследственных болезней человека. Например, субмикроскопические делеции X хромосомы описаны при хороидеремии, xLRP, и болезни Норри.
3. Моногенные мутации. Многие наследственные болезни глаз развиваются вследствие патологических изменений какого-либо одного гена. Лучше всего описаны мутации замены одного основания, которые также называют «точечными мутациями». Кардиффская база данных мутаций генов человека (Cardiff Human Gene Mutation Database) является он-лайн хранилищем информации о выявленных мутациях генов человека. Патогенные точечные мутации могут приводить к замене одной кодируемой аминокислоты на другую (миссенс-мутации). Если эти изменения вызывают нарушение функции протеина, это приводит к болезни.
Изменение одного основания, которое приводит к образованию стоп-кодона из кодона, в норме кодирующего какую-либо аминокислоту, называется нонсенс-мутация. Большинство нонсенс-мутаций вызывают уменьшение количества протеина, производимого в процессе трансляции.
После транскрипции из незрелой молекулы мРНК в процессе сплайсинга вырезаются лишние участки, и формируется зрелая мРНК. Сплайсинг — сложный процесс, в ходе которого происходит взаимодействие огромного протеинового комплекса (сплайсосомы) с молекулами мРНК. Существует огромное число мутаций — в особенности локализующихся на соединении между экзонами и нитронами или вблизи от него — вызывающих прерывание процесса сплайсинга (мутации сплайсинга).
Другие часто встречающиеся мутации ДНК, вызывающие моногенные болезни человека,— это мелкие делеции/инсерции, при которых утрачивается или вставляется до 20 пар оснований ДНК. Мутации инсерции/делеции, длиною менее трех оснований, вызывают сдвиг рамки считывания гена и образование преждевременного терминального кодона. В результате большинства таких мутаций образуется мРНК, с которой не транслируется полипептид.
в) Секвенирование ДНК. Считается, что при заболеваниях, передающихся по законам Менделя, большинство больных являются носителями одного патогенного изменения ДНК (мутации). Большинство таких мутаций находятся в пределах или вблизи кодирующих последовательностей генов, перечень которых все увеличивается.
1. Традиционное секвенирование ДНК. До недавнего времени секвенирование ДНК выполнялось по традиционной методике. Для этого проводилась амплификация коротких фрагментов каждого гена (возможно, 300-500 пар оснований) с использованием полимеразной цепной реакции. Следовательно, процесс секвенирования небольших генов проще и дешевле, чем больших генов. Для изучения десяти генов одинакового размера требуется в десять раз больше времени, чем для анализа одного гена. Такая работа стоит дорого и требует много времени. В некоторых ситуациях результаты генного анализа определяет тактику дальнейшего ведения пациента.
При xLRP у большинства пациентов имеются мутации одного из двух генов (RP2 и RPGR), поэтому традиционная методика секвенирования с использованием современных технологий оказывается достаточно простой и информативной для практического применения. Это также справедливо и для стромальных дистрофий роговицы, вызванных мутациями гена TGFBI хромосомы 5q31, так как количество мутаций, вызывающих дистрофии боуменовой мембраны (Тиля-Бенке и Рейс-Баклера), а также гранулярную и решетчатую I типа, очень невелико.
Но анализ мутаций может быть затруднен, даже если заболевание вызывается мутациями одного гена. Например, лабораторная диагностика при синдроме Коэна и синдроме Альстрема очень сложна из-за размеров и сложности генов, мутации которых вызывают эти заболевания. В случае АВСА4 (его мутации вызывает болезнь Штаргардта), содержащего 51 экзон и 6000-7000 пар оснований ДНК, секвенирование гена становится невероятно трудоемкой задачей. Кроме того, чувствительность метода выявления мутаций, в том числе известных мутаций АВСА4, значительно ниже 100%. Вследствие этого сильно снижается ценность отрицательного результата.
Наконец, для некоторых генов, в том числе и для АВСА4, в норме характерна высокая степень изменчивости, как для гена, так и для кодируемого протеина. Ответ на вопрос, является ли патогенной вариация, приводящая к замене одной аминокислоты, остается непростой задачей.
2. Высокоэффективное секвенирование ДНК. При генетически гетерогенных заболеваниях (напр. врожденная катаракта, нейрооптикопатии, arRP, синдром Usher), когда возможно наличие мутаций огромного числа генов и не отмечается преобладание мутации какого-либо одного гена, стратегия диагностики на основе традиционного секвенирования ДНК малоприменима. Некоторых успехов удалось достичь с появлением ДНК-чипов, позволяющих идентифицировать описанные ранее мутации (например, врожденный амавроз Лебера, болезнь Штаргардта), но эти методики применимы в основном у ранее обследованного контингента и их значение ограничено.
Массивное параллельное секвенирование ДНК, также называемое секвенированием следующего поколения, вероятно, сможет изменить эту ситуацию. Эти разработки позволяют секвенировать геном человека целиком, предоставляют возможность анализировать все экзоны всех генов или какой-либо части из них у любого пациента. С помощью этих технологических разработок уже удалось значительно ускорить процесс выявления неизвестных генов, мутации которых вызывают заболевания человека. Со снижением цены (согласно прогнозам, секвенирование всего человеческого генома в недалеком будущем будет стоить всего $1000), существует реальная возможность того, что крупномасштабные генетические исследования станут реальностью.
Эти исследования потребуют решения проблемы хранения огромного количества данных, поскольку такие системы выдают гигантские объемы информации. Кроме того, поскольку многие аномалии, вызывающие болезни глаз человека, представляют собой миссенс-нарушения, и так как огромное количество наших генов в норме имеют различия, проявляющиеся в замене одной аминокислоты на другую, возникает задача идентификации одного патогенного из огромного разнообразия доброкачественных вариантов, носителем которых является каждый индивидуум.
г) Генетический анализ: консультирование и этические аспекты. Генетический анализ становится все более доступным. Семьи и клиницисты могут использовать генетический анализ для подтверждения диагноза и типа наследования, и, возможно, в будущем, принять участие в исследованиях ген-специфичной терапии. Генетический анализ может иметь значительные и далеко идущие последствия для индивида и его семьи. Пациенту, намеревающемуся пройти генетическое обследование, возможно, нужно подумать о том, как он будет информировать своих родственников, в т.ч. дальних, как результаты анализа повлияют на его решение иметь детей и другие жизнеопределяющие решения, и о сопутствующих аспектах, например о медицинской страховке и страховании жизни. При направлении на генетический анализ большое значение имеют консультирование и информирование согласие.
1. Прогностическое или пресимптоматическое обследование. При поздно дебютирующих заболеваниях, для которых известен ответственный за их развитие ген (например, TIMP3 и фундус-дистрофия Сорсби), клинически здоровые индивиды с риском 50% могут согласиться пройти генетическое обследование и выяснить, являются ли они носителями. При поздно дебютирующих генетических заболеваниях, например болезни Хантингтона и синдромах предрасположенности к раку, важное значение приобретают качественные протоколы консультирования, учитывающие все «за» и «против» исследования, влияние его результатов на пациента и его жизнеопределяющие решения, психологическую поддержку при адаптации к результатам и другие аспекты, например страховку.
Принципы ведения пациентов, узнавших о своем диагнозе неизлечимой прогрессирующей потери зрения, которая повлияет на их жизненный выбор, зависимость от ухода и эмоциональное состояние, едины.
2. Обследование носителей. При рецессивных Х-сцепленных заболеваниях после выявления у пациента генетической мутации другие члены семьи могут согласиться пройти обследование на носительство. При родственных браках супруги смогут узнать, не являются ли они парой носителей. Женщины могут согласиться обследоваться на носительство генов Х-сцепленных заболеваний с целью принятия решения о рождении детей, выполнении пренатального обследования или чтобы быть более осведомленными и готовыми к развитию заболевания у будущих сыновей. Последствия этой информации для супружеской пары и поддержка, которая может потребоваться после выполнения исследования, должны рассматриваться как элементы процесса обследования.
3. Обследование детей. Показания к обследованию могут возникнуть при дебютирующих в детстве заболеваниях, когда результаты анализа повлияют на ведение пациента или решение о помощи в воспитании/образовании. Однако большое значение приобретает тщательное консультирование и подготовка родителей к таким решениям, поскольку информация о генетическом статусе и рисках может сильно повлиять на процесс воспитания ребенка. При заболеваниях, клинические проявления которых могут не проявиться до взрослого возраста, обычно рекомендуется подождать, пока пациент не повзрослеет настолько, чтобы принимать решения самостоятельно.
4. Пренатальное обследование. При наличии в семье известной генетической мутации, у супругов есть возможность провести пренатальную диагностику. Образцы хорионических ворсин (на 11 неделе) и амниоцентез (на 16 неделе) позволяют выполнить точную генетическую диагностику. Поскольку эти тесты инвазивны, имеется небольшой риск невынашивания.
Необходимо обратить внимание на причины, побуждающие индивидов проходить обследование. Решение о прерывании или сохранении беременности при положительных результатах исследования принимается индивидуально на основании личного опыта, устойчивости к стрессу (допинговых стратегий — coping strategies) и доступной поддержки. Хотя при поздно дебютирующих заболеваниях глаз пренатальное обследование проводится редко, в семьях с рано дебютирующими слепотой или синдромами множественных врожденных аномалий, как, например, болезни Лоу и Норри, целесообразно проведение пренатальной диагностики, а при выявлении патологии — прерывание беременности.
Преимплантационная генетическая диагностика включает в себя обследование эмбрионов при ЭКО перед имплантацией в матку. Такое исследование становится доступным при нескольких генетических заболеваниях глаз, но ставит перед нами новые этические проблемы, которые придется решать при консультировании.
д) Клиническое обследование. Клиническое обследование может иметь такое же значение, как и генетический лабораторный анализ. У не предъявляющих жалоб индивидов могут присутствовать незначительные изменения глаз, указывающие на их генетический статус. Следовательно, офтальмолог должен быть готов предварительно проинформировать и проконсультировать пациента перед проведением обследования на наследственные заболевания глаз, чтобы пациент владел информацией и был подготовлен на случай выявления генетических аномалий.
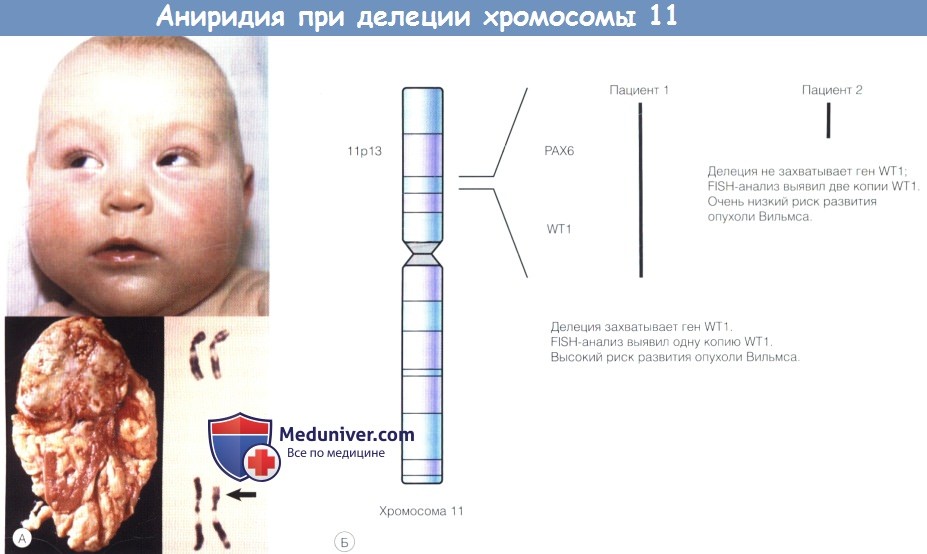
Аниридия вызвана делецией хромосомы 11.
(А) Маленький ребенок с задержкой развития, аномалиями мочеполовой системы и аниридией. Семейный анамнез по аниридии не отягощен.
В верхнем полюсе почки выявлена опухоль Вильмса. При анализе кариотипа выявлен цитогенетически видимая делеция 11р, захватывающая гены РАХ6 (аниридия) и WT1 (опухоль Вильмса).
(Б) Пациенты 1 и 2 имеют спорадическую аниридию. Хромосомный анализ не выявил патологии.
При цитогенетическом исследовании (FISH—fluorescence in situ hybridization — флуоресцентная гибридизация нуклеиновых кислот in situ) выявлено две копии гена WT1 у пациента 2,
но не у пациента 1 —пациент 1 имеет высокий риск развития опухоли Вильмса.