В развитых странах половина зарегистрированных случаев слепоты или слабовидения у детей вызывается генетическими заболеваниями, но, вероятно, это число занижено. Во многих развивающихся странах, где инвалидность детей по зрению встречается значительно чаще, генетические заболевания также являются важной группой состояний, часто вызывающих слепоту у детей.
«Генетическими» в этом контексте называются моногенные (менделевские) состояния. Многие проблемы диагностики и консультирования являются общими для всех заболеваний этой группы, что позволяет применять общий подход к ведению таких пациентов. Однако большая роль генетических факторов в развитии часто встречающейся патологии, например, была выявленная роль наследственных вариантов путей активации комплемента в патогенезе ВМД, и в наследовании нормальных количественных показателей (толщина роговицы, размер зрительного нерва), подтверждает наблюдение, что влияние молекулярных генетических факторов не ограничиваются менделевским наследованием.
Успехи в изучении наследственных заболеваний глаз являются одним из значительных достижений современной молекулярной генетики, от описания сцепления x1RP до идентификации первого гена adRP, кодирующего родопсин. Проект «Геном человека» ускорил изучение молекулярных механизмов генетических заболеваний. В настоящее время описано более 200 генных локусов и 150 генов, ответственных за моногенные заболевания сетчатки, 20 лет назад такое глубокое понимание патогенеза было недостижимо.
Геном человека распределен между 46 (23 парами, человек имеет диплоидный набор) физически различными хромосомами. Существует 22 пары аутосом, плюс две половые хромосомы: две X хромосомы у женщин, одна X и одна Y хромосома у мужчин. Хромосомы человека сильно отличаются друг от друга размером и формой, и гены, мутации которых вызывают моногенные болезни глаз, распределены между хромосомами без какой-либо закономерности.
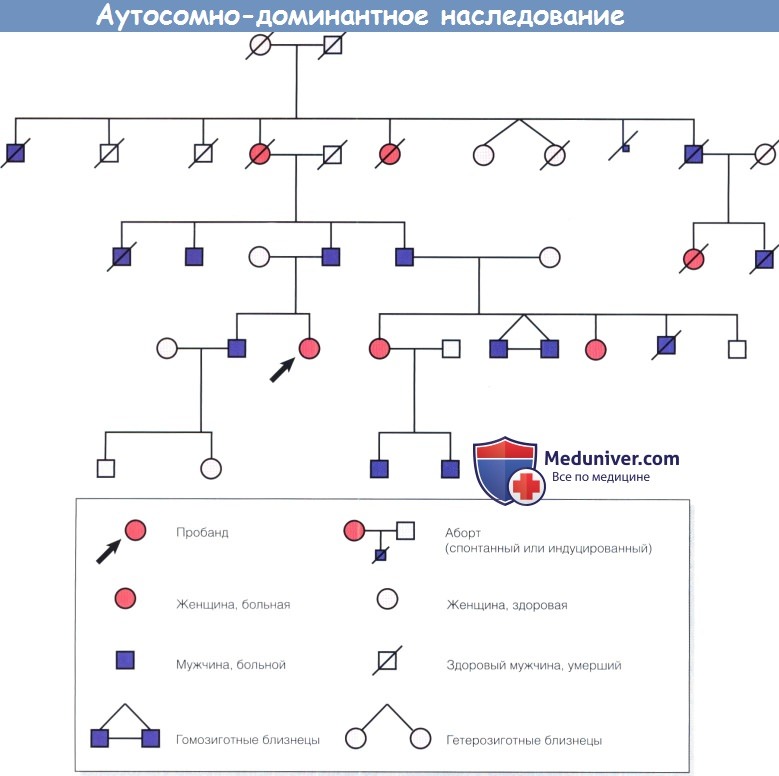
Аутосомно-доминантные (autosomal dominant — AD) болезни вызываются мутациями генов хромосом 1-22. Больные имеют одну нормальную и одну мутантную копию гена (т.е. заболевание манифестирует в гетерозиготном состоянии). В большинстве семей с аутосомно-доминантными болезнями имеются несколько поколений мужчин и женщин, пораженных в одинаковой степени, отмечается наследование болезни по мужской линии.
Шансы больных передать мутантный ген каждому потомку составляют один к двум, независимо от пола. Риск заболевания потомка здорового пациента такой же, как и в общей популяции, при условии, что здоровый пациент точно не является носителем мутантного гена.
а) Экспрессивность. Члены одной семьи, пораженные моногенной болезнью, являются носителями одной генетической аномалии. Однако проявления патологического состояния могут варьировать в широких пределах. В этом случае говорят, что заболевание или, правильнее, мутантный аллель, характеризуется вариабельной экспрессивностью.
В качестве примеров можно привести синдром Марфана, неврофиброматоз I типа и окулокутанный альбинизм, при которых глазные и экстраокулярные проявления заболевания у носителей мутации варьируют в широких пределах. Фенотипически тяжесть заболевания носителя очень мало или вообще не влияет на прогноз заболевания у его сиблинга или потомка. Это ведет к неопределенности трактовки результатов консультативного или пренатального генетического обследования. Поэтому при аутосомно-доминантном наследовании важно обследовать родителей больного ребенка с целью выявления у них легких проявлений заболевания и прогнозирования 50% риска заболевания будущих потомков.
б) Пенетрантность. При некоторых аутосомно-доминантных болезнях вероятность развития клинической картины у носителей гена ниже 100% (т.е. мутация характеризуется сниженной пенетрантностью). Следовательно, при многих заболеваниях (например, различные формы аутосомно-доминантного пигментного ретинита (AD retinitis pigmentosa — adRP) колобома или врожденная катаракта), у носителей гена признаки заболевания могут отсутствовать, но риск развития заболевания у их потомков такой же, как и у потомков больных носителей.
Это еще одна причина для обследования родителей детей, например, с колобомой или дисгенезом переднего сегмента. Доступность генетических тестов способствует правильной оценке рисков.
в) Вновь возникшие мутации. Доминантные болезни могут также возникать de novo. В этом случае семейный анамнез не отягощен, заболевание возникает вследствие копирования дефекта ДНК одного из родителей. Это часто наблюдается при анирдии или ретинобластоме. В таких случаях риск развития заболевания у будущих сиблингов гораздо ниже 50%. Эта цифра не равна нулю из-за вероятности гонадного мозаицизма (т.е. у одного родителя мутация имеется в какой-то части сперматозоидов или яйцеклеток).
Трудно определить точную природу мутации, возникшей de novo — в случаях спорадической аниридии делеция может захватить и другие соседние гены. Это наблюдается при синдроме WAGR (аббревиатура от Wilms tumor, Aniridia, Genitourinary defect, mental Retardation — опухоль Вильмса, аниридия, дефект мочеполовой системы и умственная отсталость), когда делеция вызывает развитие опухоли Вильмса, аниридию, аномалии развития мочеполовой системы и умственную отсталость. Такое явление называется синдромом соседних генов.
Поэтому пациенту со спорадической аниридией необходимо или выполнять скрининговое ультразвуковое исследование почек, или подтвердить молекулярными методами, что ген опухоли Вильмса WT1 не изменен в результате новой мутации.
При возникновении новой аутосомно-доминантной мутации риск передачи ее потомкам пораженного пациента составляет 50%. Примеры таких состояний включают в себя редкие формы врожденного амавроза Лебера (вызываемые мутациями гена CRX) и ретинобластомы (вызываемой метациями в гене RB1). Поскольку мутация RB1 может характеризоваться сниженной пенетрантностью, наличие здоровых родителей может означать, что больной ребенок является носителем мутации, возникшей de novo, или что один из родителей является носителем мутации со сниженной пенетрантностью. Генетическое обследование поможет выявить членов семьи — носителей вызывающих заболевание генов и составить прогноз.