Синдромы с краниосиностозом - методы диагностики, лечения по Европейским рекомендациям
Синдромные краниосиностозы представляют собой гетерогенную группу патологических состояний, характеризующихся связью раннего слияния нескольких черепных швов и различных врожденных пороков развития, особенно лица и конечностей. Отличительной чертой синдромных черепно-лицевых дизостозов является вовлечение как мозгового черепа (свода и основания), так и лицевого черепа (орбитальная и средняя зоны лицевого скелета). Свод черепа характеризуется множественными синостозами швов и гипоплазией верхней челюсти. До сих пор неизвестно, следует ли часто ассоциированные аномалии развития головного мозга и динамики ликвора, выявляемые при многих из этих состояний, рассматривать как первичные или вторичные проявления.
Вероятно, количество и время слияния швов могут играть прямую роль в фенотипе, по крайней мере, при некоторых из этих патологических состояний, о чем свидетельствует более часто встречающееся грыжевое выпячивание миндалин мозжечка при синдроме Крузона чем при синдроме Аперта, последний из которых характеризуется поздним слиянием ламбдовидного шва по сравнению с первым.
При этих пороках часто нарушен венозный отток из полости черепа, с развитием вторичной гипоплазии задней черепной ямки и различной степени повышения давления ликвора с расширением субарахноидального или желудочкового пространства. Существует несколько гипотез для объяснения этих изменений: венозная обструкция может быть вторичной по отношению к аномалии роста костей, расстройство может быть первично вызвано ростом диспластического основания черепа или в связи с нарушением венозного оттока у плода и нарушением нормального созревания дренажа задней ямки.
Кроме того, при некоторых этих состояниях, в связи с недостаточным развитием средней трети лица появляется экзофтальм с риском повреждения роговицы даже при небольшой травме. В дальнейшем недоразвитие дыхательных путей, в ряде случав, может вызывать изменения функции внешнего дыхания, из которых самыми тяжелыми являются ночные апноэ. Многообразное участие различных структур и черепно-лицевых функций оправдывает общий термин «фациокраниостенозы», указывающий на конкретные ассоциации костных аномалий черепа и лицевого скелета и подчеркивает трудности в их лечении.
а) Генетика краниосиностозов. Были выявлены некоторые мутации, ответственные за синдромные краниосиностозы. Обычно они включают три из четырех факторов роста, преимущественно с аутосомно-доминантным типом наследования. При синдромных синостозах один генный дефект порождает различные клинические фенотипы. Предполагается разница во взаимодействии с компонентами внеклеточной матрицы, что могло бы объяснить разнообразия фенотипов при общих дефектах гена.
б) Основные синдромы. Краткий обзор основных синдромных краниосиностозов приведен в таблице ниже.
1. Краниосиностоз при синдроме Крузона (черепно-лицевой дизостоз). Синдром Крузона — аутосомно-доминантный синдром, впервые описанный Крузоном в 1912 г. На него приходится около 4,8% случаев краниосиностозов при рождении. Распространенность была оценена как 1:25000 родов. Синдром проявляется краниосиностозом и гипоплазией лица. Фенотип краниосиностоза индивидуально отличен, но в большинстве случаев участвуют коронарные швы. Фенотип лица характерен гипертелоризмом, короткой верхней губой и относительным прогнатизмом нижней челюсти с обратным прикусом. Экзофтальм связан с ретрузией лобной области и верхних челюстей.
Эти особенности можно увидеть при рождении, но обычно они появляются в возрасте двух лет с постепенным ухудшением. Тем не менее, существуют некоторые врожденные формы, при которых гипоплазия верхней челюсти является более заметной. Больные страдают от затруднения дыхания и сильного экзофтальма, что может привести к ухудшению смыкания век. Вентрикуломегалия встречается практически всегда, иногда прогрессирующая. Мальформация Киари 1 при этом синдроме, встречается примерно в 70% случаев, может ассоциироваться с сирингомиелией и осложнять хирургическое лечение. Этот порок может быть связан с небольшим размером задней черепной ямки и преждевременным слиянием ламбдовидного шва во время первых двух лет жизни.
Ген, ответственный за синдром Крузона расположен на длинном плече хромосомы 10. Более 30 мутаций FGFR2 гена (экзон IIIа и IIIс) были идентифицированы, и они выявляются приблизительно у 60% пациентов.
Мутация в FGFR2 также может привести к синдрому Джексона-Вейсса, во многом сходному с синдромом Крузона. Тем не менее, пострадавшие субъекты также предъявляют увеличенные большие пальцы ног и тарзометаnарзальное сращение.
Мутации в FGFR3 могут привести к определенной форме синдрома Крузона, связанной с кожными аномалиями (акантокератодермия).
2. Краниосиностоз при синдроме Пфайффера (акроцефалосиндактилия тип V). Синдром был описан в 1964 г. Пфайффером. Частота синдрома Пфайффера была оценена в 1:200000. Он наследуется аутосомно-доминант-ным путем с полной пенетрантностью и варьирующей экспрессией. Синдром Пфайффера — брахицефалия, мембранная синдактилия рук и ног с увеличенными и наклоненными большими пальцами рук и ног. Могут выявляться брахидактилия, анкилоз локтевых суставов и различные висцеральные пороки развития.
В настоящее время синдром делится на три подтипа. Подтип 1 является классической и самой легкой формой, с бикоронарным синхондрозом, ведущим к брахицефалии и плоскому лицу, гипертелоризмом и слабой синдактилией с широкими большими пальцами. Подтипы 2 и 3 являются спорадическими и более тяжелыми, с выраженным экзофтальмом и аномалиями мозга, часто, но не всегда связаны с высокой летальностью. Череп в форме листа клевера характеризуется, хотя и не всегда, подтипом 2 с гидроцефалией и маленькой задней черепной ямкой и аномалией Киари 1 типа. Череп в форме листа клевера — признак очень плохого прогноза.
Синдром Пфайффера является генетически гетерогенным. Он может быть связан с мутацией в гене FGFR1 или возникать в связи с несколькими типами мутаций в FGFR2. Мутации одного и того же гена, FGFR2, таким образом, могут привести к трем типам фациокраниосиностозов, т. е. Крузона, Джексона-Вейсса и Пфайфера давая основания для предположения об участии в патогенезе и других факторов.
3. Краниосиностоз при синдроме Апера (акроцефалосиндактилии тип I). Синдром Апера является врожденным синдромом, описанным в 1906 г., связывающим фациокраниосиностоз с синдактилией на конечностях. Заболеваемость составляет примерно 1:50000 живорожденных. В большинстве случаев является спорадическим из-за отцовской de novo мутации в экзоне IIIа FGFR2 гена. Описано и аутосомно-доминантное наследование.
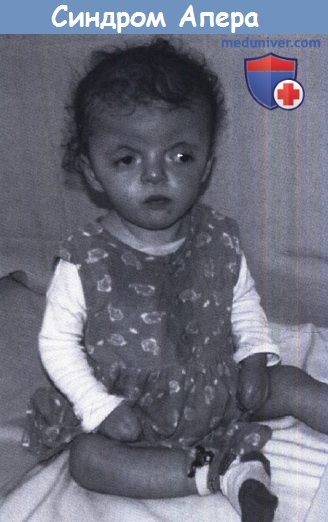
Краниостеноз является бикоронарным и затрагивает продольной шов. Лицо большое, с гипертелоризмом и экзорбитизмом, нос по типу клюва попугая, прикус перевернутый. Синдактилия может быть полной или с сохранением большого пальца и/или мизинца. Также часто наблюдается подвывих шейных позвонков. Церебральные пороки, в основном с участием мозолистого тела и лимбической структуры, могут быть выявлены с помощью МРТ. На аутопсии было найдено ненормальное развитие височной доли с дисгенезией парагиппокампальной области. Размер желудочков часто увеличен, вентрикуломегалия практически никогда не бывает прогрессирующей. Различная степень умственной отсталости связана с синдромом Апера, около одной пятой пациентов с коэффициентом интеллекта ниже 50, однако также сообщается и о людях с нормальным интеллектом.
Высокий уровень психических нарушений связан с аномалиями мозга, в том числе обонятельных-лимбико-септо-каллозальных структур, гипоплазией белого вещества, аномалиями пирамидных путей и непрогрессирующей «ассиметричной» вентрикуломегалией.
4. Краниосиностоз при синдроме Сетре-Хотцена (акроцефалосиндактилии тип III). Первые случаи были зарегистрированы Saethre в 1931 г. и Chotzen в 1932 г.. Более 40% случаев являются семейными. Передача аутосомно-доминантная с неполной пенетрантностью и варьирующей экспрессией. Наиболее типичным фенотипом является брахицефалия и недоразвитие верхней челюсти. Преждевременное слияние черепных швов обычно включает в себя коронарные швы.
Тем не менее, может быть вовлечен любой шов с асимметрией лица, необычной формой уха, с частичной синдактилией пальцев рук и ног (второй и третий пальцы, третий и четвертый пальцы с небольшими дистальными фалангами). Большой палец, как правило, увеличен, но не искривлен. Птоз, симметричный или нет, наблюдается почти во всех случаях. Этот синдром может быть чрезвычайно разнообразным в своих клинических проявлениях, таким образом, обследование членов семьи имеет первостепенное значение для выявления потенциальных носителей. Геном, ответственным за синдром Сетре-Хотцена, является TWIST, расположенный на хромосоме 7. Умственная отсталость является редкостью.
5. Краниосиностоз при синдроме Карпентера (акроцефалосиндактилии тип II). Этот синдром, аутосомно-рецессивный, крайне редкий и характеризуется акроцефалией, синдактилией мягких тканей рук, синдактилией и полидактилией ног. Некоторые авторы описывают его с ожирением и гипогонадизмом.
6. Краниосиностоз при синдроме Лежена-Мюнке. Синдром Лежена-Мюнке (Lajeunie-Muenke) характеризуется одно- или двусторонним коронарным синостозом, недоразвитием верхней челюсти, гипертелоризмом и птозом. Передача аутосомно-доминантная. У некоторых пациентов, синдром связан со скелетными аномалиями такими, как наперсткообразные фаланги среднего пальца, конические эпифизы и/или неврологические нарушения, а именно нейросенсорная потеря слуха или умственная отсталость. Несмотря на переменный фенотип, этот синдром был связан с уникальной мутацией в гене FGFR3, Pro 250-Arg.
в) Функциональные аспекты. Слияние нескольких швов, которое встречается при синдромном синостозе, приводит к худшему прогнозу интеллектуального развития, усугублению состояния и повышению внутричерепного давления, чем при моношовных синостозах. Более того, аномалии морфологии лица и лицевого роста также могут привести к недостаточной защите глаз, препятствиям дыханию, неправильному прикусу и скученности зубов, которые часто требуют конкретных процедур.
Повышение внутричерепного давления (ВЧД) является, по сути, частым для синдромного краниосиностоза. Повышенное ВЧД не только вторично по отношению к синостозу, но и встречается также в сопровождении гидроцефалии и венозных аномалий.
Нарушения зрения могут следовать за повышением ВЧД, но они также могут быть вторичными по отношению к экзофтальму, типичному при этих синдромах. Стоит отметить, что атрофия зрительного нерва и потеря зрения наблюдается в основном при синдроме Крузона.
Частота отставания умственного развития определяется типом синдрома и сопутствующими пороками развития мозга (в основном нарушения развития прозрачной перегородки). Умственная отсталость часто встречается при синдроме Апера, который представляется наиболее серьезным состоянием, и у некоторых пациентов с синдромом Пфайффера, особенно у пациентов с деформацией черепа в виде листа клевера. С другой стороны, умственная отсталость при синдроме Крузона встречается редко. В общих чертах, когнитивные функции лучше после ранней хирургической коррекции и в случае хорошей психосоциальной среды ребенка.
г) Принципы лечения синдромных краниосиностозов. Кранио-фациальная хирургия выполняет задачи как функционального, так и эстетического характера. Первый год жизни имеет первостепенное значение для лечения с целью снижения аномально повышенного внутричерепного давления и, в конечном итоге, нормализации потока ликвора. В самом деле, в первый год, мозг растет очень быстро, и результаты интеллектуального развития гораздо лучше у больных, оперированных на ранних сроках. Дополнительные преимущества раннего хирургического вмешательства включают более легкую реконструкцию костей и высокий потенциал их роста, обеспечивающий «физиологическое» заполнение возможных костных дефектов, которые следуют за хирургической коррекцией.
Классическое лечение фациокраниосиностоза включает в себя переднюю реконструкцию черепа, как первый шаг, и коррекцию лица в качестве второго шага.
Передний доступ позволяет заниматься как проблемой аномального ВЧД, так и надглазничной рецессией, следовательно, защитить глаза и зрительные функции. Лобно-орбитальное вытяжение может быть выполнено с использованием флотирующей техники или горизонтального вытяжения у детей старше 6-месячного возраста. Улучшение состояния лицевого черепа может быть достигнуто путем остеотомии по типу Le Fort III. В отдельных случаях могут выполняться фронтофациальные моноблочные перемещения с одновременной мобилизацией орбиты и лицевых костей. В настоящее время для достижения адекватного вытяжения лица предпочтительно использовать внутренние или внешние дистракторы.
Как правило, вытяжение лица должно быть отложено до тех пор, пока окончательно не сформируются зубные ряды и не возникнет стабильная окклюзия. Тем не менее, в отдельных случаях, особенно в случае нарушения дыхания, может быть необходимо раннее лицевое вытяжение. В других случаях перед хирургическим вмешательством должна быть наложена трахеостома.
Раннее расширение заднего черепного свода может также быть выполнено как первый шаг для снижения внутричерепного давления, предполагая фронтофациальную коррекцию до старшего возраста ребенка. Это может быть достигнуто, в частности, при затылочном уплощении, когда задняя ямка чрезмерно мала. Процедура обычно приводит к постепенному улучшению венозного оттока из черепа. Раннее заднее расширение может быть достигнуто как увеличением теменно-затылочного костного лоскута, так и применением пружинной краниопластики у детей с открытым лямбдовидным швом. Расширение БЗО также может быть необходимо в некоторых случаях с симптомной мальформацией Киари 1 типа.
Расширение желудочков, встречающееся при синдромном краниосиностозе также может потребовать лечения. Часто это связано с грыжей миндаликов различной степени, и расширение БЗО может стать необходимым. В других случаях, с механическим препятствием току ликвора, эндоскопическая вентрикулоцистерностомия может обеспечить необходимый контроль ВЧД. Тем не менее, в ряде случаев синдромного краниосиностоза венозный отток нарушается. Прогрессирующее открытие коллатерального венозного канала может привести к отсутствию гидроцефалии. Тем не менее, некоторым пациентам может потребоваться установка вентрикулоперитонеального шунта.
Во всех случаях требуется несколько видов хирургического вмешательства. Тесное сотрудничество между пластическим хирургом и нейрохирургом, а также детским нейроанестезиологом является обязательным условием получения хорошего функционального и косметического исхода.
Умственная отсталость у детей с синдромными краниосиностозами в зависимости от возраста на момент операции.
Распространенность непрогрессирующей вентрикуломегалии и гидроцефалии при синдромных краниосиностозах.
Виды операций при синдромных фациокраниосиностозах.
Три поколения пациентов с синдромом Крузона.
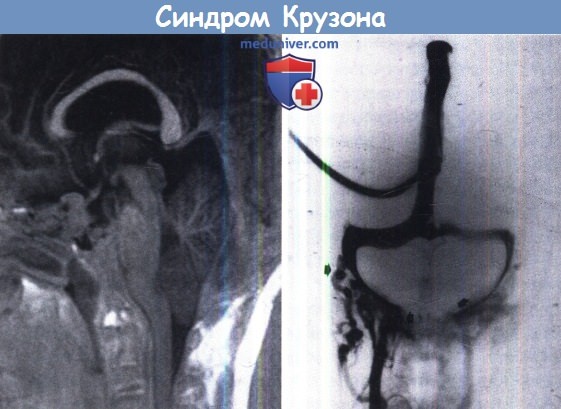
Слева: МРТ (сагиттальная проекция) мозга у ребенка с синдромом Крузона.
Обратите внимание на акроцефальную форму черепа и сопутствующие деформации церебральных структур,
расширение желудочков и каудальное смещение миндалин мозжечка (мальформация Киари I типа).
Справа: ангиография в венозной фазе. Отметьте призанки нарушения венозного оттока, сочетающегося с коллатеральными дренажами (стрелка).
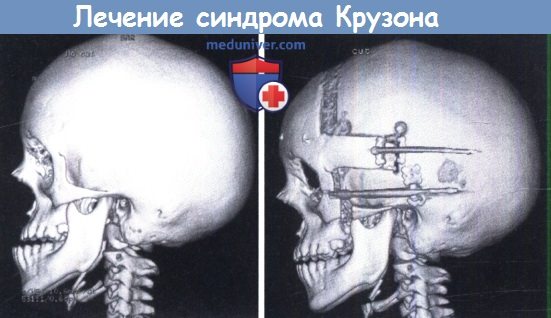
Слева: интраоперационная КТ головы у ребенка с синдромом Крузона.
Видно смещение лица кзади и обратный прикус.
Справа: тот же ребенок во время дистракции после фронто-фациального расширения.
Обратите внимание на коррекцию смещения лица.
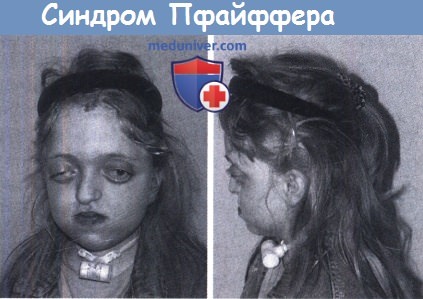
Ребенок с синдромом Пфайффера. Ухудшение дыхания вынудило к выполнению трахеостомии.
Широкий большой палец руки и огромный большой палец ноги характерны для синдрома Пфайффера.
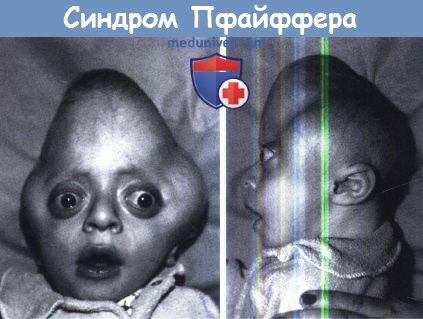
Деформация черепа в виде листа клевера.
Ребенок с синдромом Апера: типичная деформация лица в сочетании с синдактилией.
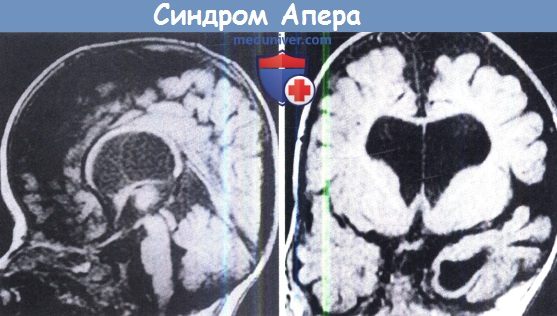
Сагиттальная (слева) и коронарная (справа) МРТ мозга у ребенка с синдромом Апера.
Отметим расширение желудочков, тонкое мозолистое тело, аномалии височной доли и отсутствие тонзиллярных выпячиваний.