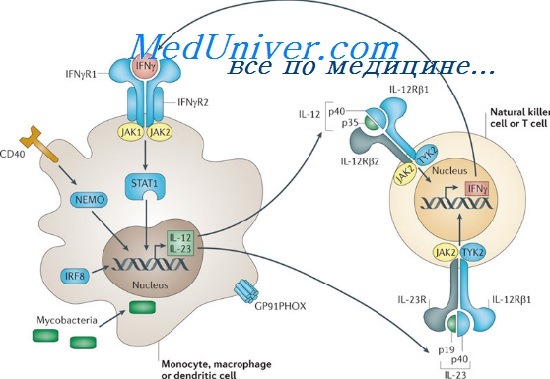
Генетика менделирующей восприимчивости к грибковой и бактериальной инфекции - mendelian susceptibility to mycobacterial disease (MSMD)
В отличие от недостаточности гликозилирования белка, происходящего при I-клеточной болезни, показано, что неожиданно высокая пропорция (приблизительно 1,5%) миссенс-мутаций, вызывающих болезнь у человека, могут быть связаны с аномально высоким N-гликозилированием, вызванным мутациями, создающими новые сайты N-гликозилирования, располагающиеся в мутантных белках.
Такие новые сайты могут привести к неправильному гликозилированию мутантного белка с патологическими последствиями, что было обнаружено у некоторых больных с редким аутосомно-рецессивным заболеванием, менделирующей восприимчивостью к грибковой и бактериальной инфекции (MSMD, от англ. mendelian susceptibility to mycobacterial disease).
Пациенты с MSMD могут иметь дефекты в любом из множества генов, включая рецепторы интерферона, регулирующие защиту от инфекций. Последствие — больные подвержены распространению условно-патогенных грибковых и бактериальных инфекций, например бациллы Кальметта-Герена (БЦЖ), используемой во всем мире, как вакцина против туберкулеза, или нетуберкулезных бактерий окружающей среды, в норме не вызывающих заболеваний.
В небольшой группе пациентов MSMD болезнь вызвана миссенс-мутацией в гене рецептора у-интерферона II типа (IFNGR2), создающей в мутантном белке IFNGR2 новые сайты N-гликозилирования. Эти новые сайты приводят к синтезу аномально большого, чрезмерно гликозилированного рецептора. Мутантные рецепторы достигают поверхности клетки, но не реагируют на у-интерферон. Поскольку удаление новой углеводной цепи восстанавливает ответную реакцию, утрата функции рецептора может быть отнесена к повышенному гликозилированию, а не к любому другому эффекту миссенс-мутаций, вызывающей патологию.
Мутации, ведущие к избыточному гликозилированию, также ведут к утрате функции белка при нескольких других моногенных заболеваниях.
Анализ базы данных мутаций генов человека показывает, что среди всех миссенс-мутаций преобладают ведущие к избыточному N-гликозилированию, указывая, что вследствие таких мутаций происходит множество наследственных болезней. Выглядит вероятным, что мутации, увеличивающие О-гликозилирование, будут также патогенны.
В отличие от этого, мутации, ведущие к снижению N-гликозилирования, в базе данных мутаций генов человека представлены мало, что, в отличие от ситуации с I-клеточной болезнью, позволяет предположить, что не все события гликозилирования критичны для функции белка.
Наконец, открытие, что удаление аномальных полисахаридов восстанавливает функцию мутантных белков IFNGR2, связываемые с MSMD, позволяет надеяться, что нарушения такого типа могут оказаться чувствительными к химиотерапии, направленной на снижение чрезмерного гликозилирования.