Врожденное отсутствие фермента галактозо-1-фосфат-уридил-трансферразы ведет к накоплению в печени токсических количеств галактозы и других метаболитов. Это заболевание с аутосомно-рецессивным типом наследования начинается в детстве с возникновения рвоты, недоедания и гепатоспленомегалии.
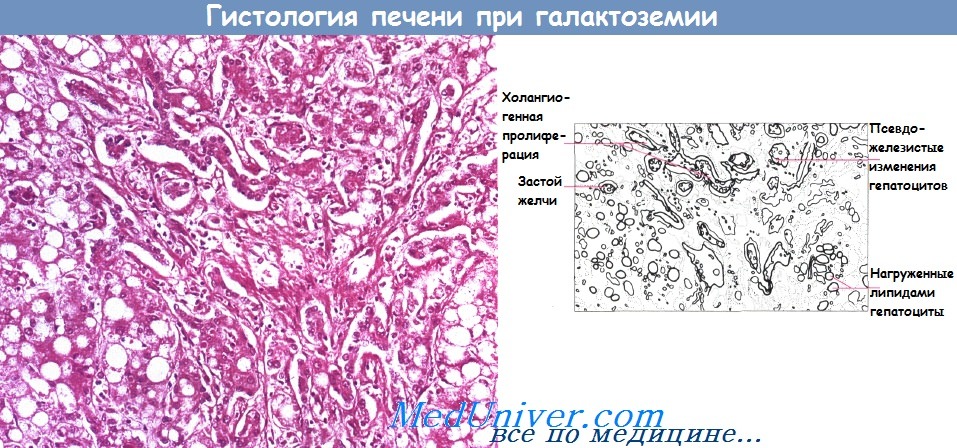
Затем развиваются катаракта, поражение мозга и макроузловой цирроз печени. Жировая дистрофия печени — ранний гистологический маркер галактоземии. Впоследствии развивается выраженные псевдожелезистые изменения гепатоцитов, уменьшающиеся с развитием цирроза.
Гистологическая картина жировой инфильтрации, псевдожелезистого повреждения гепатоцитов и пролиферации желчных протоков при галактоземии.