Митохондриальные миопатии встречаются относительно редко, но внутри этой группы болезней часто наблюдаются поражения ЦНС и нервно-мышечной системы. Разделение чисто миопатических и более комплексных форм в некоторой степени искусственно, так как митохондрии имеются в каждой клетке. Неравномерная гетероплазмия, из-за которой, вероятно, в скелетной мускулатуре могут преобладать патогенные митохондриальные мутации по сравнению с другими тканями, может также вызывать выраженное поражение преимущественно мышечной ткани и симптоматику миопатии, что может иметь и иные объяснения (DiMauro и Gurgel-Giannetti, 2005).
Изменчивые клинические проявления и сложность методов исследований, необходимых для постановки точного этиопатогенетического диагноза, превращают эти расстройства в сложную задачу для клиницистов. Тем не менее, существует несколько распознаваемых синдромов, и их генетические, биохимические, патофизиологические и клинические проявления изучаются все больше (Schapira и Cock, 1999; DiMauro и Gurgel-Giannetti, 2005; DiMauro и Hirano, 2005).
Митохондриальная дисфункция может развиться на любом этапе митохондриального метаболизма липидов и других соединений. Эти проблемы обсуждаются в отдельной статье на сайте. В данном разделе описаны только миопатии при митохондриальных болезнях, вызванных дисфункцией системы переноса электронов (дыхательной цепи).
а) Клинические проявления. Митохондриальные миопатии могут проявиться в любой период от рождения до взрослого возраста и чрезвычайно разнообразны по симптоматике, тяжести и исходу (Harding et al., 1988; DiMauro 1993). Некоторые являются составной частью комплексных энцефаломиопатий (глава 8), тогда как другие полностью или преимущественно ограничены патологией поперечнополосатой мускулатуры.
При клиническом проявлении в виде чисто миопатического процесса со слабостью преимущественно проксимальных отделов конечностей, такие симптомы, как утомляемость, миалгия, непереносимость физических нагрузок (легкая нагрузка вызывает непропорциональные тахикардию и диспноэ) и эпизодическая вызванная физической нагрузкой миоглобулинурия, хотя и не являются патогномоничными симптомами, являются ключевыми при постановке диагноза. Птоз и прогрессивная наружная офтальмоплегия — очень важные диагностические признаки (Moraes et al., 1989), так же, как и наличие кардиомиопатии.
Часто наряду с миопатией наблюдаются другие проявления мультисистемного поражения, что способствует синдромальной диагностике, например синдрома Кернса-Сейра. В исследовании Jackson et al. (1995) важными признаками расстройств дыхательной цепи явились материнское наследование, глухота и офтальмоплегия. Отсутствие прибавки в весе, пигментный ретинит, диабет, нарушения интеллекта, припадки и мозжечковая атаксия также имеют диагностическое значение. Гипотония может быть выраженной, но она имеет меньшее значение при дифференциальной диагностике.
Рваные красные мышечные волокна при синдроме Кернса-Сейра.
Обратите внимание на красные отложения—скопления митохондрий — под сарколеммой наиболее пораженных волокон.
Те же, но менее выраженные изменения начинают развиваться и в других волокнах.
б) Методы исследований. Если клинически не удается распознать имеющийся синдром, первые диагностические признаки, полученные при помощи лабораторных и инструментальных методов исследования, указывающие на патологические изменения в дыхательной цепи — это повышение уровня лактата в крови и ЦСЖ или аномалии базальных ганглиев, выявленные при лучевом исследовании (Jackson et al., 1995). При некоторых описанных синдромах, обычно не проявляющихся преимущественно миопатией (напр. MELAS, MERRF, NARP), мутации можно выявить при исследовании циркулирующих в крови лейкоцитов, таким образом, исчезает необходимость выполнения биопсии.
Даже если отсутствует выраженная слабость, гистологическое и гистохимическое исследование мышечной ткани дает указания или подтверждает диагноз митохондриального расстройства. Характерные морфологические аномалии, лучше всего видные при трехцветной окраске по Гомори, — это так называемые рваные красные мышечные волокна, возникающие из-за скопления митохондрий под сарколеммой. При электронной микроскопии можно выявить патологические митохондриальные структуры, которые могут содержать паракристаллиновые включения.
Тем не менее, рваные красные волокна при подтвержденных митохондриальных болезнях могут постоянно или временно отсутствовать. Дальнейшее обследование включает в себя гистохимическое окрашивание для оценки активности ферментов — сукцинатдегидрогеназы (СДГ), которая служит доказательством пролиферации митохондрий, и оксидазы цитохрома С (СОХ). СОХ кодируется как ядерной, так и митохондриальной ДНК (мтДНК), и мозаичный паттерн указывает на аномалию гетероплазменной мтДНК, тогда как общее снижение свидетельствует о мутации ядерной ДНК (Taylor et al., 2004).
Обычно разорванные красные мышечные волокна СОХ-негативны (Byrne et al., 1985), но могут быть и СОХ-позитивными (Taylor et al., 2004; DiMauro и Hirano, 2005). В некоторых случаях наблюдается избыток липидов и гликогена. Биохимический анализ активности комплекса дыхательной цепи может выявить дефекты в одном компоненте дыхательной цепи или нескольких комплексах, но такой анализ выполняется только в специализированных центрах. Дальнейшее уточнение проводится с помощью молекулярных генетических исследований зон митохондриальной и ядерной ДНК, ответственных не только за дыхательную цепь, но также за плотность митохондрий, их структуру и даже передвижение (DiMauro и Hirano, 2005).
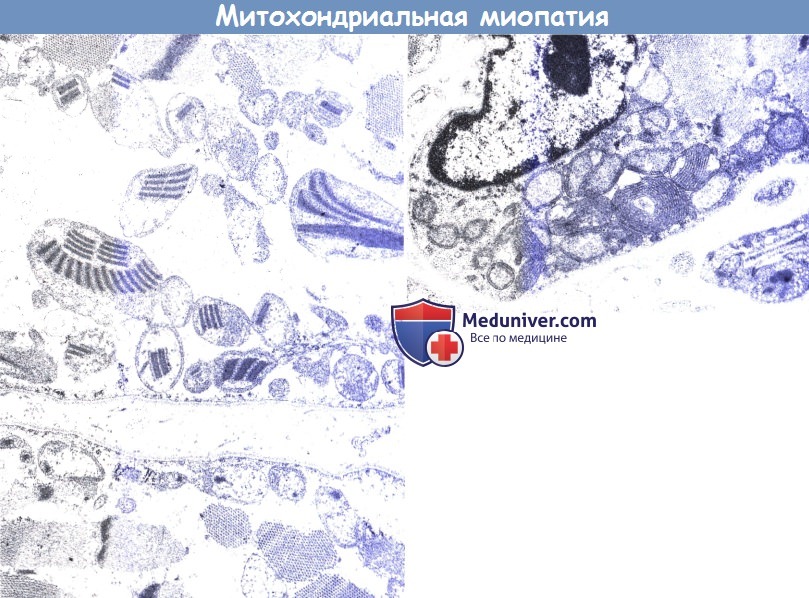
Митохондриальная миопатия (электронная микроскопия, Х28.500).
13-летний пациент с типичным анамнезом (слева). Обратите внимание на классические включения по типу «парковки» внутри митохондрии. Пятилетний пациент (справа).
Обратите внимание на пролиферацию крист внутри митохондрии. «Парковочные места» и упорядоченные паракристаллические включения в возрасте до 7-8 лет обычно не видны.
в) Клинические синдромы миопатии. Выраженная генетическая гетерогенность, отсутствие корреляции фенотип-генотип и мультисистемная природа митохондриальных расстройств свидетельствуют о том, что, хотя открыто несколько синдромов энцефалопатии с поражением мышц (например, MERRF, MELAS), существует очень мало фенотипов, характеризующихся преимущественно или исключительно миопатией. Ниже обсуждаются некоторые исключения.
г) Цитохром С оксидаза (СОХ). Одна интересная форма недостаточности СОХ проявляется тяжелым врожденным лактацидозом, слабостью и гипотонией, которые могут быть изолированными или сопровождаться поражением сердца и/или почек. Болезнь дебютирует у младенцев в возрасте от нескольких недель до трех месяцев и обычно быстро прогрессирует, вызывая дыхательную недостаточность (Zeviani et al., 1985; Darin et al., 2003). Обратимая форма недостаточности СОХ проявляется у новорожденных снижением спонтанной двигательной активности, птозом, арефлексией, повышением уровня КК и лактата сыворотки крови, но лактата в ЦСЖ не зафиксировано (Zeviani et al., 1987).
Такая же обратимость была описана при мутации гена СОХШ мтДНК, вызывающей недостаточность СОХ, которая проявляется в детстве непереносимостью физической нагрузки, генерализованной мышечной слабостью с болезненными мышечными крампи и утомляемостью (но без миоглобинурии) (Horvath et al., 2004). Nozaki et al. (1990) описали недостаточность СОХ, остро манифестирующую в возрасте семи лет, сопровождающуюся птозом, офтальмоплегией и остановкой дыхания; после терапии коэнзимом Q10 наступило выздоровление.
д) Недостаточность коэнзима Q10 (KoQ10). При манифестации недостаточности в мышцах коэнзима Q10 (убихинона) доминируют симптомы энцефалопатии, мозжечковой атаксии, также отмечается некоторая мышечная слабость (Musumeci et al., 2001). Наоборот, в некоторых случаях заболевание дебютирует симптомами миопатии: непереносимостью физической нагрузки, прогрессирующей мышечной слабостью проксимальных отделов и нагрузочной миоглобинурией. Наблюдаются разорванные мышечные волокна, накопление липидов, снижение содержания KoQ10 и снижение активности комплексов дыхательной цепи I+III и II+III (Di Giovanni et al., 2001).
Миопатия может быть изолированной (Lalani et al., 2005), но ей также могут сопутствовать симптомы энцефалопатии, например припадки или атаксия. Очень важна правильная диагностика этого расстройства, так как отмечается выраженный положительный эффект перорального приема KoQ10 (Lalani et al., 2005), хотя и не всегда (Aure et al., 2004).
е) Деплеция митохондриальной ДНК. Синдром митохондриальной деплеции (Elpeleg, 2003) часто проявляется как мультисистемное расстройство, но может быть и тканеспецифичным; миопати-ческая форма манифестирует в младенчестве или раннем детстве прогрессирующей слабостью, гипотонией, арефлексией, рано развивается дыхательная недостаточность, смерть наступает в течение первого десятилетия жизни (Mancuso et al, 2002). У некоторых пациентов были выявлены мутации гена тимидинкиназы 2 (ТК2) хромосомы 16 (Saada et al., 2001; Mancuso et al., 2002). Описаны мутации гена SULCA2, также связанные с деплецией митохондриальной ДНК и прогрессирующей мышечной слабостью, но в семьях с этой мутацией в наибольшей степени выражены симптомы мультисистемных расстройств (Elpeleg et al., 2005).
ж) Другие митохондриальные миопатии. Развитие сидеробластной анемии в подростковом возрасте может быть патогномоничным признаком синдрома, названного «миопатия и сидеробластная анемия» (MLASA); заболевание начинает манифестировать непереносимостью физической нагрузки в детском возрасте. Были выявлены мутации гена псевдоуридинсинтетазы 1 (PUS1) (Bukhovskaya et al., 2004). Манифестирующая в детстве непереносимость физических нагрузок также является ранним проявлением мутации гена цитохрома b мтДНК (Andreau et al., 1999).
Прогрессирующая наружная офтальмоплегия (ПНО) наблюдается при синдроме Кернса-Сейра как одно из проявлений мультисистемного расстройства, что в 80% случаев вызвано точечной мутацией мтДНК мт 3243 (Hirano и Pavlakis, 1994). Множественные делеции мтДНК, вызванные мутациями генов ANTI, Twinkle и POLG1, наблюдаются при наследственной форме ПНО без других системных нарушений, хотя большинство случаев манифестируют во взрослом возрасте (Agostino et al., 2003).
з) Лечение. Лечение митохондриальных болезней рассмотрено в отдельной статье на сайте.