Микроцефалию иногда определяют как уменьшение окружности головы более, чем на два стандартных отклонения ниже среднего (Barkovich et al., 2001а) или менее 3-го центиля (Haslam, 2000), однако Barkovich et al. (2001а) определили ее как «окончательную микроэнцефалию». Оба определения, очевидно, включают нормальных индивидуумов. Микрэнцефалия, с другой стороны, теоретически относится к маленьким размерам мозга (Friede, 1989), но на практике оба термина равноценны.
Все авторы допускают, что дети с умеренно маленькими головами (между 2 и 3 СО ниже среднего) часто нормальные, но статистически отмечается преобладание легкой микроцефалии среди детей с трудностями в обучении. Задержка умственного развития наиболее характерна, когда небольшие размеры головы сопровождаются задержкой развития. Однако даже пациенты с окружностью головы между 3 и 4 СО ниже среднего могут иметь нормальный интеллект.
Этиология микроцефалии разнообразна (Opitz и Holt, 1990). Многие случаи, называемые вторичной микроцефалией, являются результатом приобретенных кластических повреждений или других патологических процессов во время внутриутробного развития или позднее, в первые годы жизни, во время быстрого роста головы и мозга (Hughes и Miskin, 1986). Существенная доля случаев, несомненно, имеет первичный характер, в основном генетического происхождения. Причины подобных случаев почти неизвестны.
Недостаточная клеточная пролиферация является лишь одним из этих механизмов, потенциально способным вызывать чрезмерную гибель эмбриофетальных клеток. Генетическая микроцефалия может принадлежать к нескольким видам. Sugimoto et al. (1993) обнаружили, что 44 из 55 случаев имели генетическое происхождение, остальные были связаны с различными негенетическими ошибками развития.
Первичная микроцефалия (также называемая «истинной» микроцефалией или «microcephalia vera») наследуется по рецессивному типу (Suri, 2003). Частота варьирует в зависимости от популяции с определенным влиянием распространенности близкородственных браков. Степень микроцефалии обычно значительная (3 СО и часто 5-6 СО ниже среднего), узкий выступающий лоб и заостренное темя демонстрируют своеобразную клиническую и рентгенологическую картину. Парадоксально, при microcephalia vera отсутствуют тяжелые неврологические симптомы, но нередки гиперкинетическое поведение и расстройства тонкой двигательной координации (Accardo и Whitman, 1988).
В типичных случаях судороги не развиваются. Имеет место некоторая степень задержки умственного развития, но она обычно легкая и большинство детей могут освоить, по крайней мере, упрощенный язык. Все это становится более заметным, когда во взрослом состоянии вес мозга становится менее 500 г.
С патологической точки зрения, извилины мозга выглядят нормальными и кора истончена. Отмечается тяжелое истощение нейронов в II и III слоях (Evrard et al, 1989).
Mirocephalia vera генетически гетерогенная. Как правило, имеет место рецессивный тип наследования. Были картированы шесть локусов (МСРН1-5) и определены два гена (Suri, 2003). В трети семей, обследованных Suri, не было продемонстрировано связей, определяющих дальнейшую гетерогенность. Третий ген недавно был выявлен при похожем состоянии распространенном в секте амишей, американских меннони-тов. Ген в МСРН1 локусе кодирует белок, микроцефалии, связанный с ростом мозга. У гетерозигот ген может вызывать легкую умственную отсталость, иногда с относительно маленьким размером головы.
В других случаях микроцефалии кора мозга патологически истончена с небольшим количеством аномальных извилин. Эти случаи были названы микролиссэнцефалией (Barkovich et al., 1998) или олигирической микроцефалией (Dobyns и Barkovich, 1999). Вероятно существует несколько подтипов (Ross et al., 2001). Они клинически отличаются от первичной микроцефалии наличием глубокой задержки умственного развития, судорогами и неврологической симптоматикой, а иногда зрительной и другой патологией (Silengo et al., 1992, Kroon et al., 1996).
MPT мозга показывает различные аномалии извилин коры помимо небольших размеров мозга и плохо развитых лобных долей (Steinlin et al., 1991, Sugimoto et al., 1993, Sztriha et al., 2004). К другим редким типам мироцефалии относится мозжечковая гипоплазия (Kato et al., 1999, Ross et al„ 2001, Barth, 2003a) и синдром Неу-Лаксова (Manning et al., 2004).
Эти случаи являются частью гетерогенной группы «микроцефалии плюс». Tolmie et al. (1987) обнаружили, что только в 1 из 29 исследованных случаев имелась «microcephalia vera», тогда как в остальных клинические проявления были сложнее, включая судороги и/или спастичность. Большинство случаев были, несомненно, рецессивными (Harbord et al., 1989). Другие типы могли иметь доминантный характер наследования (Burton, 1981). Однако встречалось и множество ассоциированных аномалий, включая врожденный нефротический синдром (Nishikawa et al., 1997), короткую тощую кишку (Nezelof et al, 1976), агенезию мозолистого тела и тяжелые расстройства миграции (King et al., 1995).
Семейные случаи с внутричерепной кальцификацией, маскирующейся под внутриутробную инфекцию (синдром Айкарди-Гутиере), представляет особый интерес из-за сходства с пренатальными инфекциями, поэтому диагностика семейного состояния в этих случаях обычно упускается (Baraitser et al., 1983).
Другой конец спектра представлен редкими случаями клинически незаметной, передающейся по доминантному типу, микроцефалии (Haslam и Smith, 1979, Accardo и Whitman, 1988).
Случаи с наличием или без расстройств неврологического развития с хромосомной нестабильностью (Seemanova et al., 1985) или первичной комбинированной иммунной недостаточностью (Berthet et al., 1994) встречаются редко. Исчерпывающий перечень синдромов с микроцефалией представил Friede (1989).
Крайний вариант микроцефалии — радиальный микромозг. По Evrard et al. (1989), вес мозга в таких случаях может быть менее 16-50 г. Колонки радиального мозга почти нормальные, но их число значительно снижено. Такие случаи следует отличать от ателэнцефалии, при которой кортикальные структуры отсутствуют.
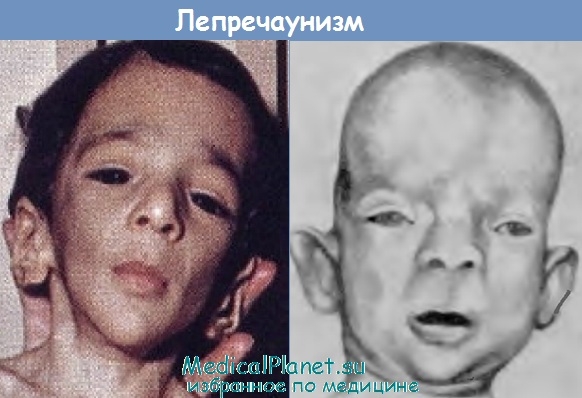
Микроцефалия при лепречаунизме
Микроцефалия также является признаком множества хромосомных аномалий, дисморфичного синдрома с умственной отсталостью и синдромов с карликовостью, такого как синдром Секкеля. Патология этих заболеваний обычно плохо изучена. Некоторые из этих синдромов имеют генетическое происхождение.
Диагноз микроцефалии прост при крайних проявлениях, но может быть сложен при легких формах. Размер головы в норме различается в семьях и индивидуально. Измерение размеров головы является неточным и обычно используемая затылочно-лобная окружность не вполне коррелирует с объемом мозга, поэтому целесообразно измерение при КТ. (Gooskens et al, 1989). Однако измерение окружности головы остается простым распространенным методом оценки размеров мозга. У детей окружность головы быстро нарастает по мере роста мозга, поэтому соответствующая кривая роста должна использоваться у новорожденных и детей раннего возраста по средней массе при рождении, а также у больных и здоровых недоношенных новорожденных (Gross et al., 1983).
У здоровых детей масса мозга утраивается в течение первого года жизни, поэтому при обследовании обязательны повторные измерения.
Дифференциальная диагностика с тотальным краниосиностозом не является проблемой. Форма черепа, экзофтальмия, нормальное развитие, признаки повышенного внутричерепного давления и рентгенография в стандартных проекциях черепа делают диагноз очевидным. Основной задачей диагноза является дифференцирование приобретенной (кластической) микроцефалии от генетических типов. Выраженный неврологический компонент, тяжелая умственная отсталость и анамнез патологических событий в пренатальном или перинатальном периодах при средней микроцефалии указывают на кластическое происхождение, а противоположные признаки относятся к генетической микроцефалии.
Однако клинические проявления в значительном объеме связаны с неопределенностью. Диагностическую ценность представляет нейровизуализация деструктивных изменений. Напротив, нормальные результаты КТ не исключают возможность приобретенного характера заболевания.
Антенатальная диагностика может быть затруднена, особенно при отсутствии сопутствующих аномалий развития. Простое измерение бипариетального диаметра недостаточно, поскольку он может быть низким у плодов с сагиттальным краниосиностозом или из-за значительного внутриутробного моделирования головки. Важны повторные измерения с особым вниманием к форме черепа.
Прогноз при микроцефалии варьирует. Некоторые дети имеют относительно хороший интеллектуальный потенциал, и длительные усилия, прилагаемые к их обучению, ненапрасны. Специальное обучение неизбежно, хотя в исключительных случаях преуспевают дети даже с окружностью головы менее 6 СО ниже среднего (личное наблюдение).