Макроцефалия, мегалэнцефалия - клиника, диагностика. Отличия макроцефалии от гидроцефалии
Макроцефалия, как и микроцефалия, определяется размерами окружности головы, произвольно ограниченными двумя стандартными отклонениями выше возрастных норм. При таком статистическом критерии под определение попадают как нормальные индивидуумы, так и целый ряд разнообразных и несоотносимых случаев. Большинство случаев макроцефалии не связано с патологией развития мозга, но здесь будет обсуждаться в связи с большими размерами головы как основного и заметного признака. Lorber и Priestley (1981) обследовали 510 детей с окружностью головы более 98-го центиля.
У 75% из них наблюдалась гидроцефалия и повышенное внутричерепное давление, у 3% — специфические синдромы и у 20% отмечалась первичная мегалэнцефалия с нормальным давлением. Только у 13% из последней группы развилась умственная отсталость или неврологические отклонения. Основные диагностические критерии — гидроцефалия или перицеребральные скопления, поскольку они делают необходимым немедленное лечение.
Соотношение мальчиков и девочек 4:1, а у 50% пациентов макроцефалия отмечается в семейном анамнезе.
Макроцефалия представлена двумя основными группами: мегалэнцефалия и гидроцефалия с перицеребральным скоплением. Последняя группа описана в отдельной статье на сайте.
Мегалэнцефалия. Под мегалэнцефалией подразумевается увеличение массы мозга. Некоторые авторы (Friede, 1989) отделяют пациентов с большими размерами головы, но нормальным процессом развития и функционирования нервной системы, от случаев «истинной» мегалэнцефалии, т.е. существенного увеличения мозга по сравнению с возрастной нормой. Только последняя группа принадлежит к расстройствам развития мозга. Это различие несколько преувеличено, так как большая голова — и даже тяжелый мозг при аутопсии — нередко встречается у нормальных лиц или в сочетании с заболеваниями, такими как нейрофиброматоз, без каких-либо признаков мозговой дисфункции.
Предметом описания в данной главе будет истинная мегалэнцефалия с чрезмерными размерами мозга, связанными с патологией развития, что вызывает как увеличение числа невральных элементов, так и невральных и глиальных, или нарушение миграции и организации (Friede 1989); оба типа аномалии часто сосуществуют. Во многих случаях также выявляют гигантские патологические клетки, напоминающие таковые при туберозном склерозе и свидетельствующие о нарушении клеточной пролиферации. Причиной могут быть и иные патологические процессы, к примеру, патологическое накопление или рост отсуствующих в норме не-невральных элементов, как при болезни Александера. Структура извилин в таком мозге часто изменена.
Даже при строгом определении мегалэнцефалия включает несколько отличающихся патологических и клинических форм. Она может быть частью специфических синдромов, таких как синдром Сотоса или многочисленных нейрокожных синдромов, таких как макроцефалия-мраморная кожа (Giuliano et al., 2004), или может быть изолированной. Механизм мегалэнцефалии в настоящее время плохо понятен. Нарушена регуляция клеточной пролиферации субопухолевого (гамартоматозного) происхождения, либо диффузного, либо более локализованного, что в некоторых случаях трудно дифференцировать от ганглиоглиом. Повышенный уровень инсулиноподобного фактора роста (ИФРII) был выявлен в мозге новорожденных с макроцефалией вместе с сильно нарушенным развитием неокортекса (Schoenle et al., 1986).
Клинические проявления крайне различаются, зачастую они могут отсутствовать совсем. У большинства пациентов отмечают задержку умственного развития и у многих — судороги и диффузные неврологические симптомы.
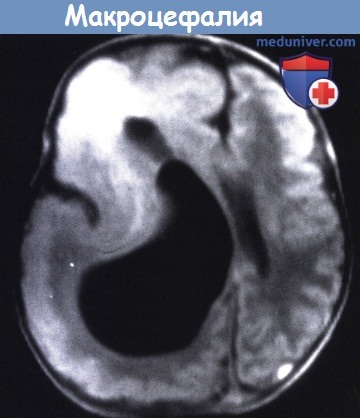
Т1 - взвешенное изображение МРТ пациента в возрасте пяти месяцев.
Большое правое полушарие, с патологически утолщенной корой и несколькими бороздами.
Значительно увеличенный желудочек и аномальный сигнал от лобного белого вещества.
Дифференциальный диагноз может быть трудным. Гидроцефалия и внутричерепные скопления должны быть исключены, поскольку требуют хирургического лечения. Дифференцировка мегалэнцефалии от больших размеров головы у детей с нормальным или практически нормальным развитием нервной системы обычно не вызывает затруднений. Однако такие дети составляют большую гетерогенную группу. У большинства из них, вероятно, большие размеры головы генетически детерминированы. Lorber и Priestley (1981) обнаружили высокую частоту макроцефалии у родителей, особенно отцов таких детей.
De Myer (1972) и Day и Schutt (1979) также предположили доминантный тип передачи «простой» макроцефалии. Некоторые из этих макроцефалических детей имели специфические нарушения способности к обучению, превышающие показатели детей с нормальным размером головы. Скорость увеличения размеров головы особенно высока у детей до четырех месяцев жизни (De Myer, 1972, Lorber и Priestley, 1981). На этой стадии часто возникают подозрения на гидроцефалию и, если темпы прироста быстро не снижаются, показана рентгенография. При отсутствии какой-либо патологии развития нервной системы и большого размера головы в семейном анамнезе не требуется ничего, кроме внимательного наблюдения. Однако необходимо исключить возможность пресимптоматической глутаровой ацидурии (Hoffmann et al., 1991) или другой органической ацидурии, часто сопровождающих макроцефалию.
У детей с долихоцефалией (синостоз сагиттального шва) важную роль играет форма черепа, потому что при выраженном удлинении черепа окружность головы у таких пациентов часто 3 СО и выше среднего, но сама по себе форма черепа вызывает подозрения. У маленьких детей с перинатальными заболеваниями или более старших, имеющих проблемы в питании, «догоняющий» рост временно осложняет диагностику (Sher и Brown, 1975). Нейровизуализацию необходимо выполнить в случаях выраженных расходящихся изгибов головы или при наличии симптомов повышенного внутричерепного давления.
Лечения мегалэнцефалии не существует, а в большинстве случаев в нем нет необходимости.