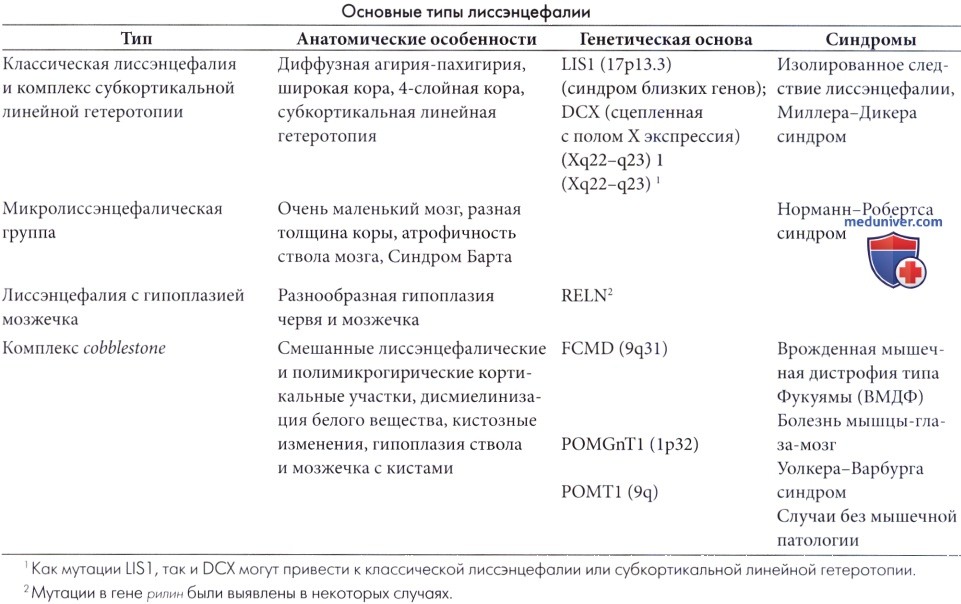
Определение комплекс cobblestone пришло на смену прежнему названию «лиссэнцефалии типа 2». В этой группе мальформаций кора имеет зернистую поверхность и полимикрогирический вид нередко наблюдается вместе с участками без извилин. Механизм образования первоначально заключается в избыточной миграции нейронов за пределы дефектной глиальной пограничной мембраны (Williams et al., 1984, Dobyns et al., 1996) в субпиальное пространство (Haltia et al., 1997). Таким образом, это состояние совершенно отлично от классической лиссэнцефалии. Помимо этого, вовлечение мышц присутствует практически во всех заболеваниях этой группы, включающей синдром Уолкера-Варбурга, болезнь мышц-глаз-мозга и врожденную мышечную дистрофию типа Фукуямы (Barkovich et al., 1998).
• Синдром Уолкера-Варбурга (Cormand et al., 2001) — сложный синдром, состоящий из изменений коры в виде «булыжной мостовой» (часто с участками полимикрогирии), зрительных аномалий, пороков развития мозжечка и тяжелой мышечной дистрофии. Кора имеет вид двойного кортикального слоя в связи с наличием субкортикальных гетеротопических островков, которые могут быть обширными. Оболочки мозга утолщены, млечного вида в результате массивной мезенхимальной пролиферации, особенно вокруг ствола. Мозжечок уменьшен с отсутствием червя. Пирамидальный тракт обычно отсутствует.
В 75% случаев развивается гидроцефалия, обычно из-за фиброза патологических оболочек, но также имеются сообщения о случаях со стенозом водопровода (Bordarier et al., 1984). Часто присутствует слияние фронтальных полюсов и молекулярных поверхностей мозжечковой пластинки. Микроскопически определяется полное нарушение кортикальной архитектоники. Кортикальная пластинка состоит из разной толщины плохо ориентированных клеток, разделенных трабекулами глиомезенхимальной ткани до мозговых оболочек. Множественные островки гетеротопического серого вещества расположены параллельно кортикальной поверхности, отделенные от вышележащих слоев коры тонкой прослойкой белого вещества, где тангенциально проходят тонкостенные сосуды. Фиброглиальная ткань изолирует клубочковые или трабекулярные образования из нейроцитов (Takashima et al., 1987). В мозжечке отмечается полная дезорганизация. Червь обычно утрачен, и на МРТ визуализируются многочисленные округлые кисты (D’Amico et al., 2006).
Синдром Уолкера-Варбурга является генетическим расстройством, передающимся по аутосомно-рецессивному типу (Dobyns et al., 1985,1989). Имеются указания на связь синдрома с мутацией РОМТ1 гена на хромосоме 1р32-34 (Cormandet al., 1999, Beltran-Valero de Bernabe et al., 2002, Balci et al., 2005). Beltran-Valero de Bernabe et al. (2004) описали случаи, связанные с мутацией гена фукутина с аналогичной клинической картиной и весьма вероятной большей гетерогенностью. Тем не менее, в большинстве случаев мутации не выявлены (Currier et al., 2005).
Среди клинических проявлений (Dobyns et al., 1985, Dobyns и Leventer, 2003) выделяют тяжелые неврологические дисфункции с рождения; патологию глаз с дисплазией сетчатки во всех случаях, а также микрофтальмию и пороки развития переднего сегмента (аномалия Питера, катаракты, персистирующее первичное стекловидное тело) у большинства; наличие у некоторых пациентов заднего цефалоцеле или большого заднего родничка. Новорожденные с тяжелыми неврологическими нарушениями и гидроцефалией в сочетании с патологией глаз имеют высокий риск наличия лиссэнцефалии II типа, относительно распространенной причины генетической гидроцефалии.
На МРТ кора выглядит как умеренно утолщенная, с неравномерной или с похожей на посыпанную галькой поверхностью с серо-белой границей (van der Knaap et al., 1997). Ствол мозга атрофичен, червь мозжечка имеет малые размеры или апластичен. Часто присутствуют небольшие, округлые кортикальные кисты в мозжечке (Aida et al., 1994). Синдром Уолкера-Варбурга редко сопровождается развитием приведенных больших пальцев и стенозом водопровода (Bordarier et al., 1984), и его следует отличать от Х-сцепленного синдрома Беккера из-за разного генетического компонента. Пренатальная диагностика возможна на основании гидроцефалии, цефалоцеле и патологии глаз (микрофтальмия и/или ретинальная отслойка) (Rhodes et al., 1992) и может подтверждаться при генетическом исследовании (Gasser et al., 1998).
Вовлечение мышечной системы является неотъемлимой частью случаев лиссэнцефалии II типа (Barkovich, 1998), хотя зарегистрированы случаи с нормальным развитием глаз и мышц (Dobyns et al., 1996). Распространенный некроз волокон определяется во всех мышцах, напоминая картину при тяжелых мышечных дистрофиях. Вероятно, цереброокуло-мышечный синдром (ЦОМС) тесно связан или идентичен синдрому Уолкера-Варбурга.
• Болезнь мышц-глаз-мозга. При этом заболевании, описанном Santavuori et al. (1989,1998), мозговая дисфункция обычно менее выраженная. Патология глаз в основном состоит из тяжелой миопии, но может также включать и осложнения, такие как глаукома и недостаточность зрительной функции (Pihko et al., 1995, Haltia et al., 1997). Желудочки расширены до умеренной степени, а перегородка обычно отсутствует. Заболевание манифестирует с рождения, но, очевидно, прогрессирует, что видно по выравниванию на нейро-ЭРГ после периода новорожденности. У 3/4 пациентов отмечаются высоко амплитудные вызванные потенциалы на вспышку. Была выявлена мутация POMGnT1 гена, находящегося на хромосоме 1р34-р32 (Cormand et al., 1999), но также была обнаружена гетерогенность.
• Врожденная мышечная дистрофия типа Фукуямы с неврологической точки зрения является менее тяжелым проявлением группы комплекса cobblestone, хотя отмечается тяжелая задержка умственного развития и часто глазные аномалии. В клинических проявлениях доминируют мышечные нарушения. Это расстройство наследуется по рецессивному типу и вызывается мутацией гена FTCD, который кодирует белок фукутин, расположенный на хромосоме 9q31.
Комплекс по типу «булыжной мостовой» (тип II лиссэнцефалии).
Т2-взвешенная МРТ: утолщенная гладкая кора. В подлежащей узкой белой полосе могут присутствовать множественные мелкие кисты.
Обратите внимание на массивную гидроцефалию и отсутствие червя мозжечка.
• Глионевральные гетеротопии представляются минимальными формами подобных аномалий в субпиальном пространстве. Они характерны для порочно развитого мозга, но также встречаются в мозге нормальных индивидуумов (Dambska et al., 1986, Hirano et al., 1992). Эти повреждения состоят из разных по размеру и форме узелков, иногда слоя астроцитов и нейронов, покрывающих молекулярный слой. Нодулярная кортикальная дисплазия, также называемая status verrucosus, состоит из описанных кортикальных деформаций, представляющих собой полукруглые выступы, 1-2 мм в диаметре, над поверхностью коры (Friede, 1989, Iida et al., 1994). Они составляют ядро радиальных миелинизированных волокон, которые проникают в поверхностные слои и расходятся в разные стороны подобно фонтану, вызывая утолщение верхних слоев. В ограниченном количестве узелки, вероятно, бессимптомны.
Подобные деформации в большем количестве присутствуют в мозге пациентов с глютаровой ацидурией типа 2 (Goodman и Frerman, 1984). Совместно с другими аномалиями они являются характерными патологическими признаками плодного алкогольного синдрома.
Прогноз при синдроме cobblestone неблагоприятный, особенно при типе Уолкера-Варбурга. Дети с наиболее выраженной патологией гибнут через несколько недель или месяцев, редкие пациенты живут несколько лет. Пациентам с гидроцефалией необходимо выполнить шунтирование, чтобы избежать значительного роста размеров головы, но в конечном итоге прогноз остается неутешительным. Тем не менее, исход при болезни мышц-глаз-мозга и дистрофии типа Фукуямы менее серьезный. При этом могут наблюдаться спектр тяжести от летальных случаев до врожденной мышечной дистрофии или даже дистрофии мышц поясов конечностей с нормальным мозгом и познавательной функцией (D’Amico et al., 2006).