Более того, появляется ряд порочных кругов, когда токсический отек легких и гипоксемия стимулируют гипоксические нарушения проницаемости мембран; поражение почек способствует дополнительной задержке жидкости в организме (стимулируется отек) и шлаков (нарастает токсемия); поражение печени с подавлением ее детоксикационной функции также углубляет токсемию; токсическая миокардиопатия усугубляет органные нарушения микроциркуляции; токсическая энцефалопатия ведет и к мозговым расстройствам, а освобождающиеся нейропептиды стимулируют нейрогенный отек легких. Именно такая "суммация" поражений при полиорганной недостаточности и определяет крайне высокую летальность - до 80%. Этот синдром полиорганной недостаточности отражает биологическую катастрофу, вид биологического суицида, возникающую при широком круге клинических ситуаций.
Поражения эндотелия легочных капилляров, помимо развития интерстициального отека, приводят также к нарушениям микроциркуляции и микротромбозам, что ведет к появлению очагов ишемического поражения легочной паренхимы и последующим деструкциям. Альвеолярный отек прекращает доступ кислорода к интерстицию, что при наличии местной ишемии и анаэробной микрофлоры ведет к гангрене легкого.
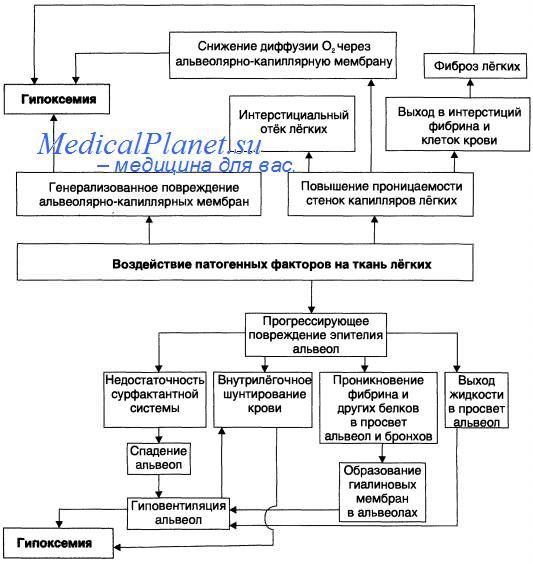
Интерстициальный и альвеолярный токсический отек легких блокирует газообмен на уровне альвеол вследствие расширения аэрогематического барьера (альвеоло-капиллярной мембраны). Это приводит к тяжелой и труднокорригируемой паренхиматозной дыхательной недостаточности, являющейся ведущим фактором танатогенеза.
Примерно такой же механизм развития респираторного дистресс-синдрома при септическом и ожоговом шоке, других видах эндотоксемии. При травматическом шоке существенный вклад на общем фоне эндотоксемии имеет жировая эмболия. Однако при этом имеется в виду не столько факт попадания в циркуляцию свободного жира из зон тканевой деструкции (что, конечно, имеет место), сколько нарушение суспензионного состояния липидов и формирование жировых глобул уже в сосудистом русле. Это активизирует липазу и в результате липолиза резко возрастает концентрация свободных жирных кислот и лизофосфатидов, обладающих выраженной мембранотропной активностью.
Патогенез респираторного дистресс синдрома взрослых (РДСВ)
При тяжелых травмах и, главным образом, при синдроме раздавливания, продолжительной ишемизации тканей и развитии аутолиза формируются чрезвычайно токсичные продукты тканевого распада, миоглобин и свободный гемоглобин (вследствие гемолиза), которые в наибольшей степени оказывают повреждающее воздействие на пути своего выведения - на паренхиму и функцию почек, что часто вызывает потребность в гемодиализе.
Токсичные продукты, циркулирующие в крови, оказывают повреждающее воздействие не только на эндотелий стенки сосудов, но и на ингредиенты самой крови, главным образом, на ее клетки. Нарушения проницаемости, механических и электростатических свойств мембран эритроцитов способствуют их агрегированию (сладжу) и еще большим расстройствам реологии крови и микроциркуляции. Возбуждение мембран лейкоцитов способствует возрастанию их адгезивных свойств и задержке в микрососудах (синдром краевого стояния лейкоцитов). Активация тромбоцитов также способствует повышению их адгезивности, возникновению микроагрегатов, которые становятся как бы ядрами для последующего формирования каскада реакций ДВС-синдрома, стимулирующего и микротромбозы и кровотечения.
Таким образом, респираторный дистресс-синдром является вторичным токсическим поражением респираторной паренхимы, возникающим при заболеваниях не только легких, но и при целом ряде других патологических состояний, имеющих общие патогенетические механизмы. Главным из них является токсическое нарушение проницаемости клеточных мембран.
Специальные исследования, проведенные еще в 1970-е годы, выявили нарушение активности сурфактанта при развитии шоковых легких. Сур-фактант, уменьшая поверхностное натяжение в альвеолах и обеспечивая тем самым их стабильность на выдохе, снижает и гидростатическое давление в легочных капиллярах, предотвращая транссудацию жидкости из них. Таким образом, отсутствие сурфактанта приводит как к ателектазу, так и к отеку легких. Главным действующим началом сурфактанта является фосфолишщ дипальмитил-фосфатадил холин, но существуют и белковые его компоненты, т.е. сурфактант представляет собой липо-протеид, синтез которого происходит в альвеолоцитах II типа.
Существует несколько попыток объяснить снижение активности сурфактанта. В частности, считается, что жидкость и белок, поступающие в альвеолу при отеке, дезорганизуют слой сурфактанта, смывают его. Однако возможна и прямая ингибиция сурфактанта под воздействием каких-то токсичных субстанций, среди которых выделяются свободные жирные кислоты. Гистохимические исследования показали, что уже через 2 часа после начата геморрагического шока наступают изменения поверхностно-активной пленки альвеол, ее фрагментация.