Подагра проявляется гиперурикемией, мочекислым нефролитиазом и острым воспалительным артритом. Подагрический артрит развивается в результате отложения кристаллов мононатрия урата, которое приводит к воспалительному процессу в суставах и окружающих тканях. Чаще всего поражается один сустав, как правило, пястно-фаланговый сустав большого пальца стопы.
Тофусы (отложения кристаллов мононатрия урата) могут появляться в проекции мест прикрепления сухожилий к костям в области локтевых, коленных суставов и стоп или в области завитка ушной раковины. Первичная подагра развивается, как правило, у мужчин среднего возраста на фоне снижения почечной экскреции мочевой кислоты, избыточного поступления пуринов с пищей, избыточного употребления алкоголя или фруктозы или сочетания указанных факторов.
Подагра возникает при любых состояниях, приводящих к снижению клиренса мочевой кислоты — на фоне терапии ЗНО, обезвоживания, лактатацидоза, кетоацидоза, голодания, применения диуретиков и почечной недостаточности. Уровни мочевой кислоты могут повышаться при избыточном потреблении пуринов, алкоголя или фруктозы. У большинства пациентов биохим. этиология подагры остается неизвестной. Считается, что основой является генетический полиморфизм, прежде всего в отношении транспортеров мочевой кислоты.
В редких случаях причина подагры — избыточная продукция пуринов, связанная с несколькими генетическими нарушениями, обсуждаемыми ниже. Вторичная подагра развивается в результате др. заболевания, сопровождающегося быстрым разрушением тканей или обновлением клеток, которые приводят к повышенной продукции или сниженной экскреции мочевой кислоты или на фоне некоторых типов медикаментозной терапии. Напр., на фоне применения диуретиков уменьшается объем плазмы, что может провоцировать приступ подагры.
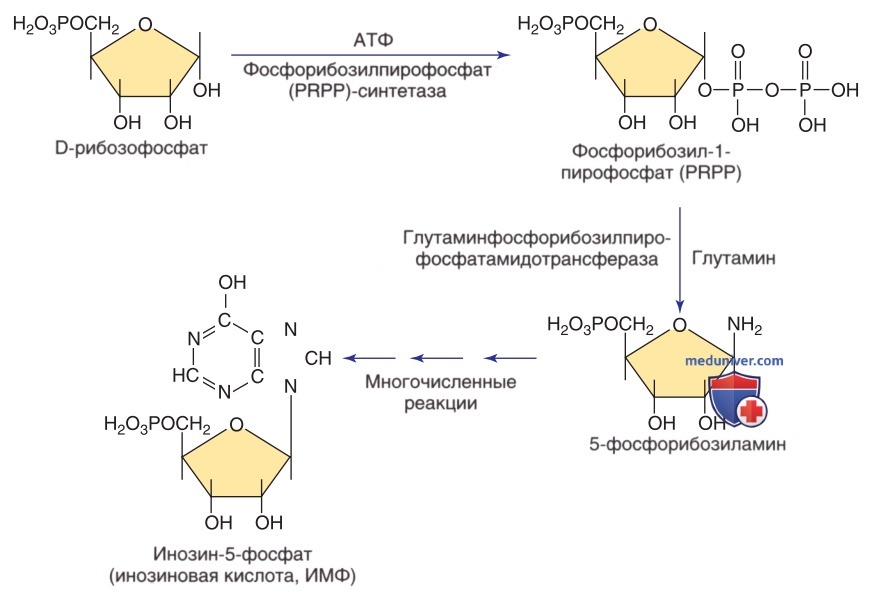
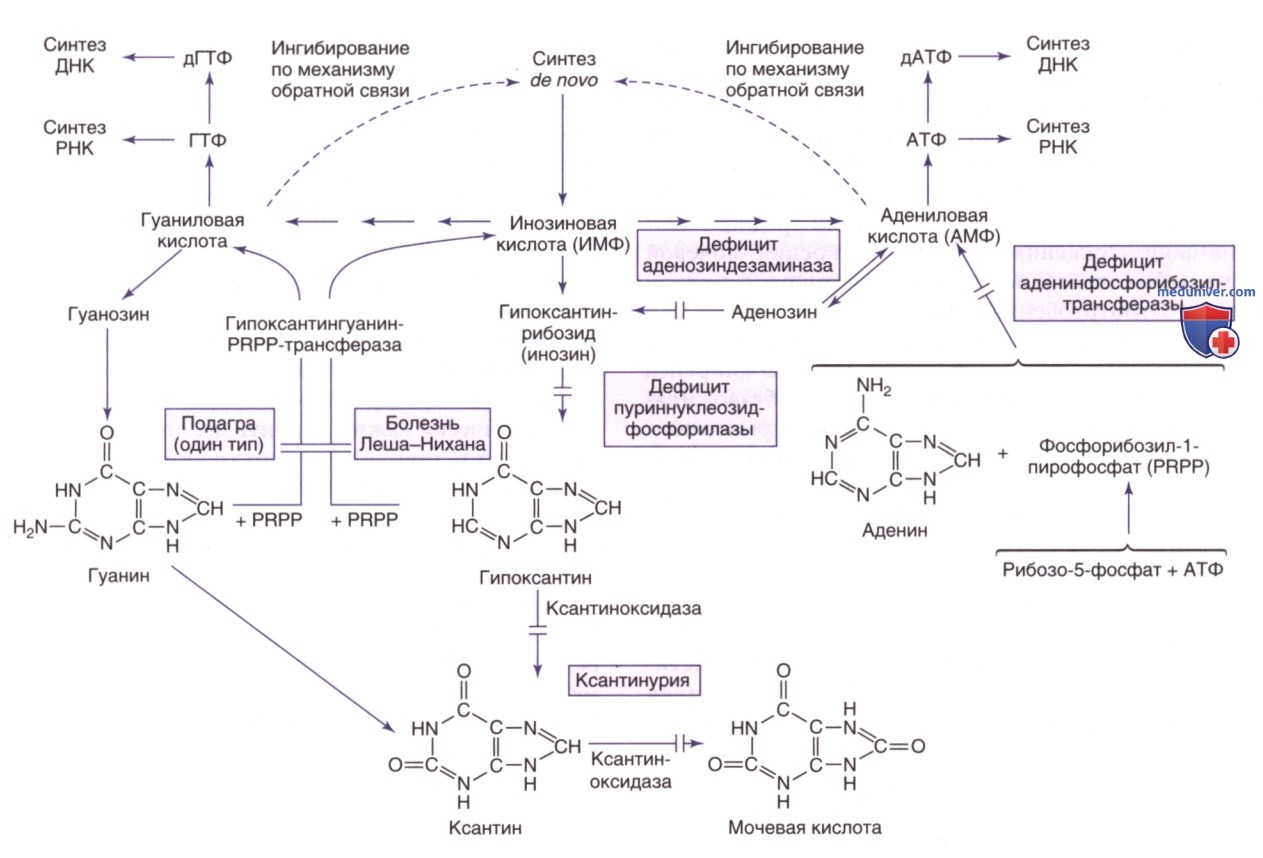
Пути метаболизма и утилизации пуринов. PRPP — фосфорибозилпирофосфат
Подагра, возникающая на фоне избыточной продукции эндогенных пуринов, связана с наследственными нарушениями трех разных ферментов, приводящими к гиперурикемии. К указанным нарушениям относятся расстройства спектра дефицита ГГФРТ (от тяжелого дефицита, или синдрома Леша-Нихана, до частичного дефицита ГГФРТ), две формы суперактивности фосфорибозилпиро-фосфатсинтетазы и гликогеноз типа I (дефицит Г-6-ФД).
При первых двух расстройствах основой гиперурикемии является избыточная продукция пуриновых нуклеотидов и мочевой кислоты, тогда как третье развивается в результате избыточной продукции мочевой кислоты и сниженной почечной экскреции уратов. Гликогенозы типов III, V и VII сопровождаются гиперурикемией, провоцируемой ФН. Эта гиперурикемия развивается в результате быстрой утилизации АТФ и отсутствии эффективного восстановления ее запасов во время выполнения физических упражнений.
Ювенильная подагра возникает в результате недостаточного выведения пуринов. Использовавшийся ранее термин «ювенильная гиперурикемическая нефропатия» заменили более новым — АуД «тубулоинтерстициальная болезнь почек». Термин «АуД тубулоинтерстициальная-уромодулин-ассоциированная болезнь почек» применяют для обозначения медуллярной кистозной болезни почек 2-го типа, ген располагается в хромосоме 16р11.2. Заболевание возникает в результате мутаций гена, кодирующего уромодулин.
Кроме того, к генам, отвечающим за развитие семейной гиперурикемической нефропатии, относят те, которые кодируют ренин и ядерный фактор гепатоцитов 1β. В отличие от трех наследственных нарушений метаболизма пуринов, которые являются Х-сцепленными гликогенозами с рецессивным типом наследования, пиримидиновые нарушения относятся к АуД. Семейная ювенильная гиперурикемическая нефропатия сопровождается сниженной фракционной экскрецией мочевой кислоты.
В типичных случаях первые признаки заболевания проявляются, начиная с пубертатного периода до 30 лет, однако зарегистрированы и случаи манифестации в раннем детском возрасте. Заболевание характеризуется началом в раннем возрасте, гиперурикемией, подагрой, семейными заболеваниями почек и низкими показателями клиренса уратов относительно СКФ. Подагра может возникать как у мужчин, так и у женщин и нередко сопровождается быстрым ухудшением функции почек, которое может привести к летальному исходу, несмотря на раннее установление диагноза и начало терапии.
При диагностировании семейной гиперурикемической нефропатии критически важно выявить др. членов семьи с гиперурикемией и, при наличии показаний, начать терапию до появления у них симптомов. Это позволит предотвратить развитие нефропатии.
а) Генетика. Причиной семейной ювенильной нефропатии-2 (HNFJ2; 613092) является мутация в гене, кодирующем ренин (REN; 179820), который локализуется на хромосоме 1q32. Ген локализуется в хромосоме 2р22.1-р21. АуД-тубулоинтерстициальная болезнь почек связана с мутациями в гене, кодирующем муцин (MUC1). Мутация в гене, кодирующем уромодулин, прослеживается до хромосомы 16.
б) Лечение. Для лечения гиперурикемии применяют комбинацию аллопуринола или фебуксостата (ингибиторы ксантиноксидазы) — с целью снижения продукции мочевой кислоты, пробенецид — для повышения клиренса мочевой кислоты у пациентов с нормальной функцией почек и употребление жидкости в больших количествах — с целью снижения концентрации мочевой кислоты. Рекомендуют придерживаться диеты с низким содержанием пуринов, снижение МТ, а также сокращение потребления алкоголя и фруктозы (фруктоза снижает клиренс уратов и ускоряет распад АТФ до мочевой кислоты).