а) Дефицит миоаденилатдезаминазы (дефицит мышечной аденозинмонофосфатдезаминазы). Миоаденилатдезаминаза — мышечноспецифическая изоформа фермента АМФ-дезаминаза, проявляющая активность в скелетных мышцах. При ФН дезаминирование АМФ приводит к повышению уровней ИМФ и аммония пропорционально мышечной работе.
Известны две формы дефицита миоаденилатдезаминазы: врожденная (первичная) форма, которая м.б. бессимптомной или сопровождаться судорогами или миалгией при выполнении упражнений, и вторичная форма, которая может сопровождаться др. нейромышечными или ревматологическими нарушениями.
1. Клинические проявления. Клинические проявления, как правило, включают изолированную мышечную слабость, утомляемость, миалгии после умеренной или интенсивной нагрузки, а также судороги. На фоне миалгии могут отмечаться повышенные уровни креатинкиназы (уровень) и определяемые отклонения на ЭМГ. Признаки атрофии мышц или гист. изменения биоптатов отсутствуют.
Заболевание может начинаться в 8 мес. При этом в ~25% случаев диагноз устанавливают в возрасте от 2 до 12 лет. Дефект фермента обнаружен у бессимптомных членов семьи. Вторичные формы дефицита мышечной АМФ-дезаминазы обнаружены при болезни Верднига-Хоффмана, синдроме Кугельберга-Веландера, полиневропатиях и амиотрофическом боковом склерозе. Метаболические нарушения включают цикл пуриновых нуклеотидов.
Как показано на рис. ниже, ферментами, участвующими в этом цикле, являются АМФ-дезаминаза, S-AMP-синтетаза и S-AMP-лиаза. Предполагается, что мышечная дисфункция при дефиците АМФ-дезаминазы связана с нарушением продукции энергии во время сокращения мышцы. Неизвестно, каким образом у людей с дефицитом этого фермента могут отсутствовать симптомы. Предполагают, что кроме дисфункции мышц мутация АМФ-дезаминазы печени приводит к избыточной продукции мочевой кислоты и является причиной первичной подагры.
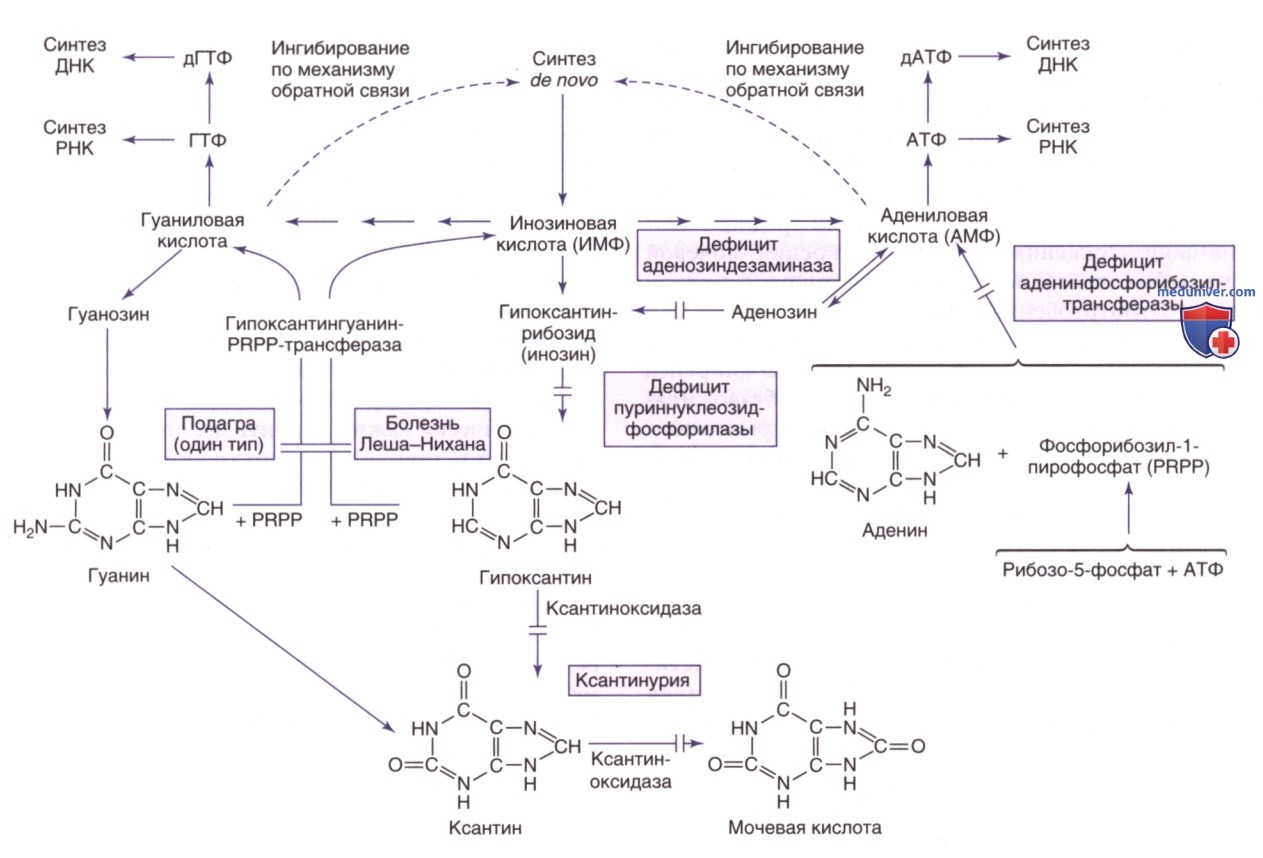
Пути метаболизма и утилизации пуринов. PRPP — фосфорибозилпирофосфат
2. Генетика. Врожденная форма заболевания передается АуР-путем. AMPD1 — ген, ответственный за кодирование мышечной АМФ-дезаминазы, — локализуется на коротком плече хромосомы 1 (1р13-21).
По данным популяционных исследований, мутантный аллель часто обнаруживают у представителей европеоидной расы, однако в результате альтернативного сплайсинга гена мутация может удаляться, и фермент функционирует нормально. В связи с этим скрининговое обследование на заболевание проводят методом ишемического теста на предплечье. Повышение уровня аммиака в плазме венозной крови после ФН, наблюдавшееся у лиц без данного нарушения, при дефиците АМФ-дезаминазы отсутствует.
3. Лабораторные признаки. Окончательный диагноз устанавливают по результатам гистохимического или биохимического исследования мышечных биоптатов. Первичную форму отличают по уровням ферментов <2% с незначительным уровнем иммунопреципитируемых ферментов или их отсутствием. Пациентам, страдающим данным нарушением, рекомендуют соблюдать осторожность при ФН, поскольку они могут привести к рабдомиолизу и миоглобинурии.
4. Лечение. Хотя методов лечения, позволяющих полностью излечить пациентов с дефицитом миоаденилатдезаминазы, не зарегистрировано, предполагают, что увеличение скорости восстановления пула АТФ может оказаться эффективным. Показано, что основанная на этом подходе терапия рибозой (2-60 мг/сут внутрь в несколько приемов) или ксилитолом, который превращается в рибозу, у одних пациентов повышает показатели выносливости и силы мышц, а у др. она неэффективна.
Применение генетического подхода м.б. целесообразно в будущем при врожденной форме заболевания, тогда как при вторичной форме важную роль играет лечение основного заболевания.
б) Дефицит аденозиндезаминазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
в) Дефицит пуриннуклеозидфосфорилазы. См. отдельную статью на сайте - просим Вас пользоваться формой поиска по сайту выше.
г) Дефицит ксантиноксидоредуктазы (наследственная ксантинурия/дефицит кофактора молибдена). Ксантин оксидоредуктаза (КОР) представляет собой каталитический фермент на финальной стадии катаболического пути пурина, окисляющий гипоксантин до ксантина и ксантин до мочевой кислоты. Поскольку КОР существует в двух формах: ксантиндегидрогеназа и ксантиноксидаза, то нарушение называют дефицитом ксантин-дегидрогеназы / ксантиноксидазы.
Ксантин, непосредственный предшественник мочевой кислоты, менее растворим в моче, чем мочевая кислота, а в результате дефицита фермента развивается ксантинурия. Дефицит КОР может возникать в изолированной форме (ксантинурия типа I), в комбинированной форме, включающей дефицит КОР и альдегидоксидазы (ксантинурия типа II), или множественный дефицит КОР, альдегидоксидазы и сульфитоксидазы (дефицит кофактора молибдена).
Все три формы приводят к почти полному замещению мочевой кислоты гипоксантином и ксантином в моче, при этом уровень мочевой кислоты в плазме становится очень низким или неопределяемым.
Пациенты с изолированной формой м.б. бессимптомными или иметь легкие симптомы. Почечные камни часто не видны на рентгенограмме, но они являются риском повреждения почек и могут появиться в любом возрасте. В таком случае у пациентов появляются боли в пояснице или развивается почечная недостаточность. Клиническая картина ксантинурии типа II аналогична типу I, но для пациентов также характерен дефицит альдегидоксидазы, который не имеет явных клинических признаков.
Дефицит кофактора молибдена возникает из-за наследственного дефицита синтазы кофактора молибдена, которая влияет на все три молибдофермента. Эта патология, так же как и изолированный дефицит сульфитоксидазы, обычно проявляется проблемами с кормлением новорожденных, неонатальными судорогами, повышенным или пониженным мышечным тонусом, вывихом хрусталика глаза, тяжелой умственной отсталостью и смертью в раннем детстве. В более легких случаях наблюдается только вывих хрусталика.
1. Генетика. Все три типа ксантинурии наследуются комплексно как АуР-признаки. Тип I является результатом мутаций человеческого гена XDH, расположенного в хромосоме 2р22. Ксантинурия типа II возникает в результате мутаций гена сульфуразы кофактора молибдена, расположенного в хромосоме 18q12.2; этот фермент завершает синтез кофактора молибдена, необходимого для активности КОР и альдегидоксидазы.
Ксантинурия типа III (дефицит КОР, альдегидоксидазы и сульфитоксидазы) может возникать в результате функциональных мутаций в любом из трех генов: MOCS1 (кодирует два фермента для синтеза предшественника посредством бицистронного транскрипта), MOCS2 (кодирует молибдоптеринсинтазу) или GPHN (кодирует гефирин), расположенные в 6p21.2, 5q11.2 и 14q23.3 соответственно.
2. Лабораторные признаки. Первоначально диагноз ставится путем измерения концентрации мочевой кислоты в плазме и/или моче. Содержание мочевой кислоты в плазме очень низкое, либо она полностью отсутствует (<1 мг/дл). В моче концентрация мочевой кислоты понижена, ее заменяют ксантин и гипоксантин. Пациентов типа II можно отличить по отсутствию в моче метил-2-пиридонкарбоксамида, продукта распада никотинамида (ниацина) под воздействием альдегидоксидазы.
С др. стороны, пациентов типа II можно отличить от пациентов типа I по их неспособности окислить тестовую дозу аллопуринола до оксипуринола через альдегидоксидазу. Дефицит кофактора молибдена отличается избыточной экскрецией с мочой сульфита и др. серосодержащих метаболитов, таких как сульфоцистеин.
Ферментный анализ КОР обычно не назначают, поскольку он требует биопсии тонкой кишки или печени (единственных тканей человека, содержащих значительные количества фермента). Измерение сульфитоксидазы и синтазы кофактора молибдена можно провести в печени и фибробластах. Для подтверждения диагноза может использоваться молекулярно-генетический анализ, заключающийся в поиске функциональных мутаций среди трех групп генов.
3. Лечение. Хотя изолированный дефицит обычно является доброкачественным, рекомендуется лечебная диета с низким содержанием пуринов и фруктозы (что снижает распад АТФ до ксантина) с повышенным потреблением жидкости. Аллопуринол не рекомендуется. Прогноз дефицита кофактора молибдена ранее был крайне неблагоприятным, однако испытания циклического пираноптерина монофосфата у пациентов с дефектом гена MOCS1 продемонстрировали многообещающие результаты.