Мукополисахаридозы — это группа наследственных прогрессирующих заболеваний, возникающих в результате мутации генов, кодирующих лизосомальные ферменты, которые необходимы для разложения гликозаминогликанов (ГАГ) (мукополисахариды).
ГАГ — это длинноцепочечные сложные углеводы, состоящие из уроновых кислот, аминосахаров и нейтральных сахаров. К числу основных ГАГ относят хондроитин-4-сульфат, хондроитин-6-сульфат, гепарансульфат, дерматансульфат, кератансульфат и гиалуронан. Эти в-ва синтезируются и связываются (за исключением гиалуронана) с белками, образуя протеогликаны, которые являются главными компонентами основного в-ва соединительной ткани, а также ядерных и клеточных мембран. Разложение протеогликанов начинается с протеолитической деградации ядерного белка, за которой следует постепенная деградация фрагмента ГАГ.
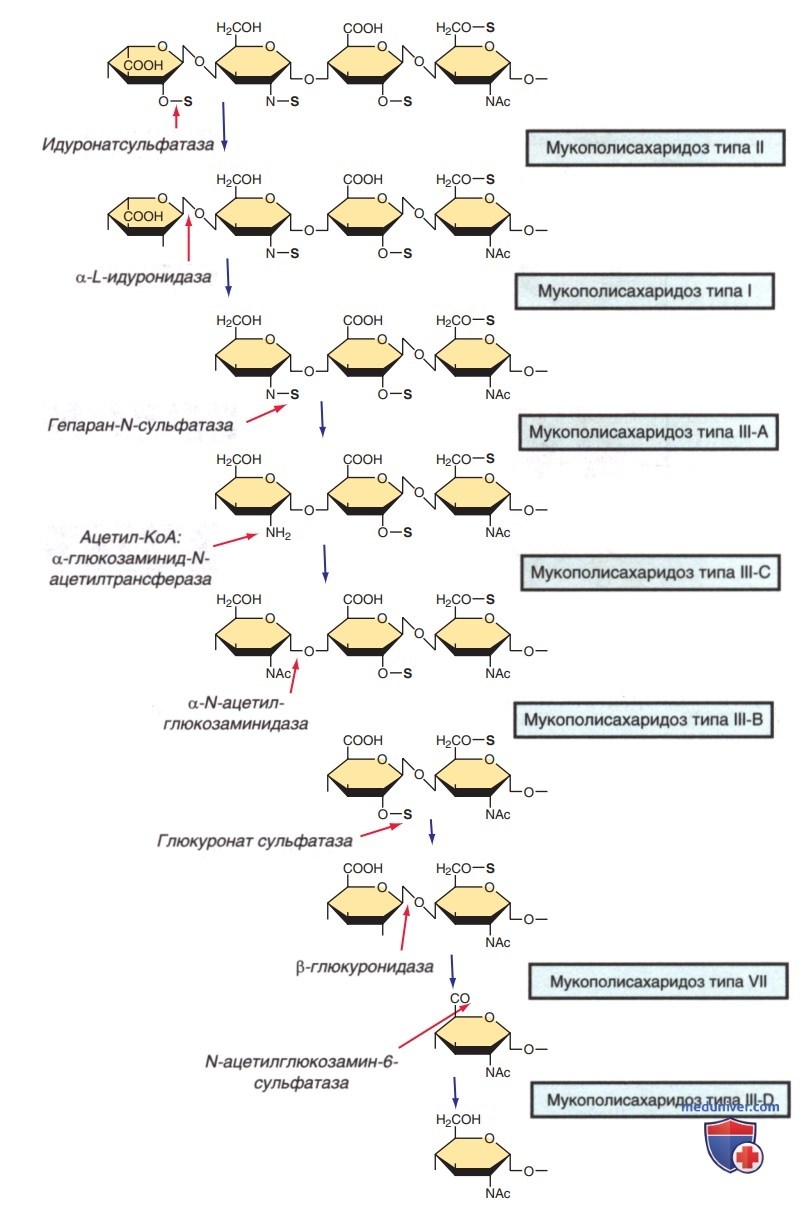
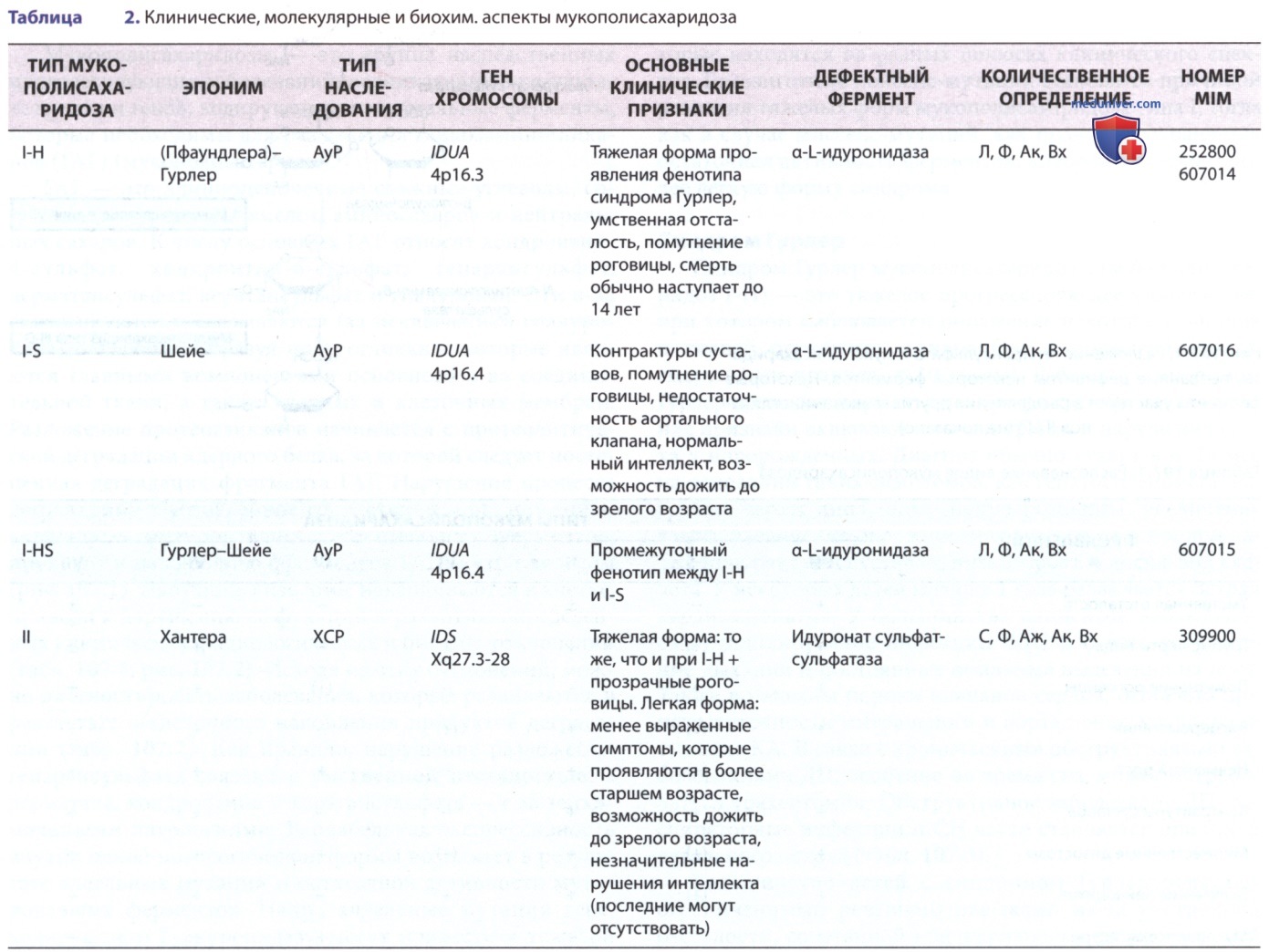
Нарушение процесса деградации, обусловленное отсутствием или снижением активности мутировавших лизосомальных ферментов, приводит к накоплению фрагментов ГАГ внутри лизосом (рис. 1). Набухшие лизосомы накапливаются в клетке, приводя к нарушению ее функций и развитию определенных клинических, радиологических и биохим. отклонений (табл. 1, рис. 2). Исходя из этих отклонений, можно диагностировать заболевания, которые развиваются в результате в/клеточного накопления продуктов деградации (табл. 2). Как правило, нарушение разложения гепарансульфата связано с умственной отсталостью, а дерматана, хондроитина и кератансульфата — с мезенхимальными патологиями.
Рисунок 1. Разложение гепарансульфата и мукополисахаридозы, вызванные дефицитом некоторых ферментов. Некоторые ферменты участвуют в расщеплении других гликозаминогликанов (ГАГ) (не показано)
Вариабельная экспрессивность внутри одной нозологической формы возникает в результате аллельных мутаций и остаточной активности мутировавших ферментов. Напр., аллельные мутации гена, кодирующего L-идуронидазу, могут привести к тяжелой болезни Гурлер (синдрому Гурлер) с летальным исходом в раннем возрасте или к ее более мягкому варианту — болезни Шейе (синдрому Шейе), которая проявляется ограничением подвижности суставов, незначительными деформациями скелета и помутнением роговицы.
Мукополисахаридозы являются АуР-заболеваниями, за исключением болезни Хантера (синдрома Хантера), которая принадлежит к числу Х-сцепленных рецессивных генетических заболеваний. Общая заболеваемость мукополисахаридозами среди родившихся детей колеблется от 1,2:100 000 (США) до 16,9:100 000 (Саудовская Аравия). В США наиболее распространен подтип мукополисахаридоз-Ш, за ним следуют подтипы мукополисахаридоз-I и мукополисахаридоз-II.
Нозологические формы:
а) Мукополисахаридоз I типа. Мукополисахаридоз типа I возникает в результате мутаций гена IUA, расположенного на хромосоме в локусе 4р16.3 и кодирующего a-L-идуронидазу. Анализ мутаций выявил два основных аллеля — W402X и Q70X, на которые приходится более половины аллельных мутаций у пациентов европеоидной расы с мукополисахаридозом типа I. Мутации, в результате которых появляются стоп-кодоны, приводящие к отсутствию функционального фермента (нулевые аллели), а также гомозиготности или сложной гетерозиготности, вызывают синдром Гурлер. Др. мутации встречаются только у одного или нескольких человек.
Дефицит a-L-идуронидазы лежит в основе широкого ряда клинических проявлений, от тяжелого синдрома Гурлер до его более мягкого варианта синдрома Шейе, которые находятся на разных полюсах клинического спектра. Гомозиготные нонсенс-мутации становятся причиной развития тяжелых форм мукополисахаридоза типа I, тогда как в случае миссенс-мутаций, как правило, сохраняется остаточная активность ферментов, которая объясняет более легкую форму синдрома.
1. Синдром Гурлер. Синдром Гурлер мукополисахаридоз-I (мукополисахаридоз I-H) — это тяжелое прогрессирующее заболевание, при котором наблюдается поражение некоторых органов и тканей организма, приводящее к преждевременной смерти (как правило, к 10 годам). Ребенок с синдромом Гурлер выглядит нормальным при рождении, однако ранние признаки включают паховые грыжи и нарушения слуха у новорожденных. Диагноз обычно ставят в 6-24 мес на основании таких симптомов, как гепатоспленомегалия, грубые черты лица, помутнение роговицы, увеличение языка, увеличение окружности головы за счет лобной части, контрактуры суставов, низкий рост и дисплазия скелета. У некоторых детей младше 1 года развивается острая кардиомиопатия.
У большинства пациентов наблюдаются рецидивирующие инфекции ВДП и отиты, затруднение дыхания и постоянные обильные выделения из носа. Также возможны пороки клапанов сердца, особенно при недостаточности митрального и аортального клапанов, и сужение КА. В связи с хроническими обструктивными заболеваниями ДП, особенно во время сна, может потребоваться трахеотомия. Обструктивное заболевание ДП, респираторные инфекции и СН часто становятся причиной летального исхода (табл. 3).
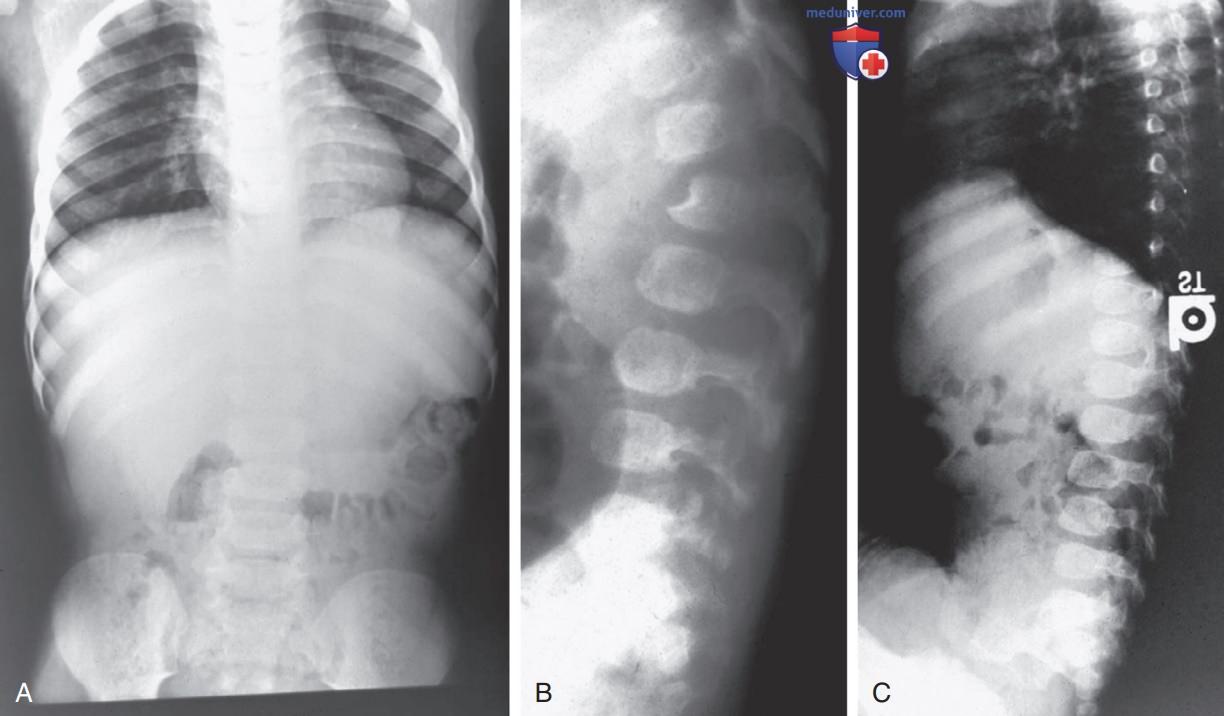
Большинство детей с синдромом Гурлер обладают ограниченными речевыми навыками из-за умственной отсталости, сочетанной кондуктивной и нейросенсорной тугоухостью и увеличением языка. Также наблюдается прогрессирующее увеличение желудочков ГМ с повышением ВЧД, возникающее в результате сообщающейся гидроцефалии. Часто встречаются помутнение роговицы, глаукома и дегенерация сетчатки. На рентгенограмме видна характерная дисплазия скелета, известная как множественные дизостозы (рис. 3, 4). Первые рентгенологические симптомы включают толстые ребра (веслообразные) и овоидные тела позвонков.
Рисунок 3. Множественные дизостозы: А — синдром Санфилиппо, возраст пациента 4 года; расширение ребер; B — синдром Санфилиппо, возраст пациента 4 года; незрелая яйцевидная конфигурация тел позвонков; C — синдром Гурлер, возраст пациента 18 мес.; передне-верхняя гипоплазия 1-го поясничного позвонка (L1) в форме крючка
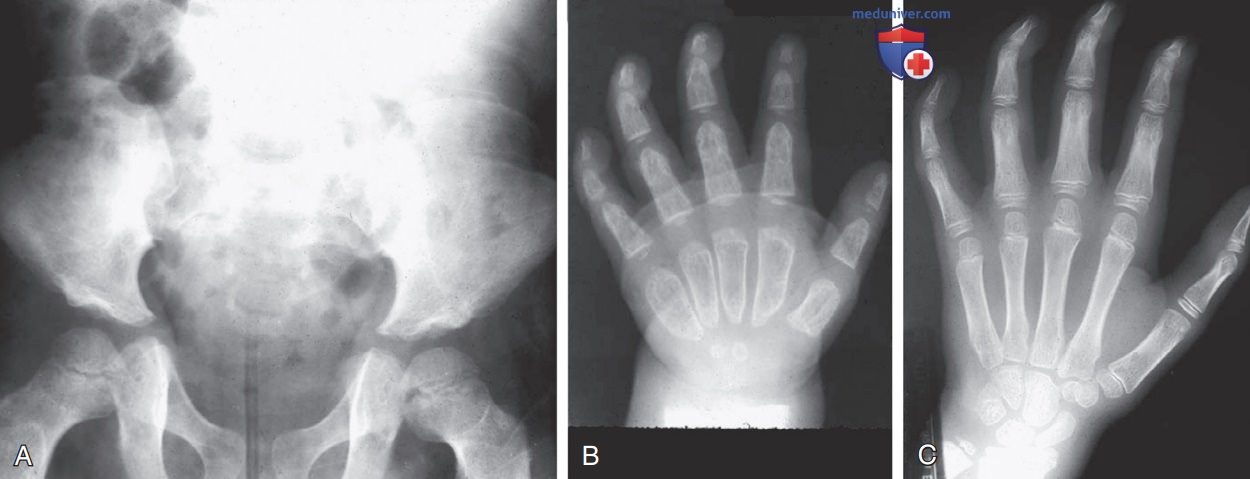
Рисунок 4. Множественные дизостозы: А — мукополисахаридоз типа l-Н, возраст пациента 10 лет. Из-за гипопластичности подвздошные кости приобретают треугольную форму наряду с неглубокими вертлужными впадинами. Вальгусная деформация бедренной кости; В — мукополисахаридоз l-Н, возраст пациента 4 года. Пястные кости и фаланги аномально укороченные, расширенные и деформированные с проксимальным расположением пястных костей и пулевидными фалангами. Крупные рыхлые трабекулы при тонком кортикальном слое кости; С — мукополисахаридоз I-S, возраст пациента 13 лет. Кости запястья маленькие, поэтому пальцы имеют V-образную форму. Хорошо смоделированные короткие трубчатые кости. Сгибание средней и дистальной фаланг II—V вызвано контрактурами суставов
К другим деформациям скелета (в дополнение к показанным на рисунках) относят увеличенные диафизы длинных костей с крупными рыхлыми трабекулами и неравномерно расположенными метафизами и эпифизами. По мере прогрессирования заболевания развивается макроцефалия, которая сопровождается утолщением свода черепа, преждевременным закрытием лямбдовидного и сагиттального швов, неглубокими орбитами, увеличенной J-образной формой турецкого седла и аномальным расстоянием между зубами с зубными кистами.
2. Синдром Гурлер-Шейе. По своему клиническому фенотипу синдром Гурлер-Шейе мукополисахаридоз-I (мукополисахаридоз I-H/S) занимает промежуточное положение между синдромом Гурлер и синдромом Шейе и характеризуется прогрессирующими соматическими расстройствами, включая множественные дизостозы и незначительные нарушения интеллекта (или может отсутствовать). Как правило, симптомы начинают проявляться в возрасте от 3 до 8 лет. Характерные признаки: скафоцефалия, макроцефалия, запавшая переносица, пухлые губы, помутнение роговицы, микрогнатия, умеренный гипертрихоз, утолщение кожных покровов, вальгусные деформации голеней незначительно выражены, умеренно ограничена подвижность в суставах, определяются дизостозы, бочкообразная ГК, кифосколиоз, гиперлордоз.
Отмечается задержка темпов психоречевого развития, позже появляется глубокая деменция*. Обычно такие пациенты доживают до зрелого возраста. К числу осложнений относят СН и обструкцию ВДП. У некоторых пациентов наблюдается спондилолистез, который может привести к компрессии спинного мозга.
P.S. * КР Союза педиатров России, Общества медицинских генетиков, «Мукополисахаридоз I», 2019 г.
3. Синдром Шейе. Синдром Шейе мукополисахаридоз-I (мукополисахаридоз I-S) представляет собой сравнительно легкую форму заболевания, характеризующуюся контрактурами суставов, недостаточностью аортального клапана, помутнением роговицы и легкими множественными дизостозами. Первые заметные симптомы обычно появляются после 5 лет, а диагноз ставят в возрасте от 10 до 20 лет.
Пациенты с синдромом Шейе обладают нормальным интеллектом и ростом, гиперстенического телосложения с сильно развитой мускулатурой, черты лица грубые, характерен широкий рот с пухлыми губами, плоская переносица, широко расставленные глаза, гипоплазированная нижняя челюсть, макроглоссия и гиперплазия десен, короткая шея, имеют серьезные поражения суставов и нарушения зрения в виде неравномерного помутнения роговицы. В дальнейшем, обычно после 30 лет, развивается глаукома, пигментная дистрофия сетчатки, редко — отек диска зрительного нерва. Часто наблюдается синдром запястного канала (туннельный синдром), редко — врожденный щелкающий I палец (болезнь Нотта).
Офтальмологические симптомы включают помутнение роговицы, глаукому и дегенерацию сетчатки. У некоторых пациентов при хронической ОДП возникает СОАС и требуется проведение трахеотомии. Довольно часто встречается недостаточность аортального клапана, которая в некоторых случаях требует проведения операции по его замене.
б) Мукополисахаридоз II типа:
1. Синдром Хантера (мукополисахаридоз-II) — это Х-сцепленное заболевание, вызванное дефицитом идуронат-2-сульфатазы. Ген IDS картирован в локусе Xq28. Точечные мутации гена IDS обнаружены приблизительно у 80% пациентов с мукополисахаридозом-II. У остальных пациентов были обнаружены серьезные нарушения в виде делеции или перестройки гена IDS, которые обычно приводят к развитию более тяжелого клинического фенотипа. Будучи Х-сцепленным рецессивным заболеванием, синдром Хантера почти всегда проявляется только у мужчин. Однако иногда он встречается и у женщин из-за искажения инактивации Х-хромосомы, несущей нормальный ген.
Выраженная молекулярная гетерогенность объясняет широкий спектр клинических проявлений синдрома Хантера. У пациентов с тяжелой формой мукополисахаридоза-II имеются симптомы, сходные с симптомами синдрома Гурлер, за исключением помутнения роговицы, а также более медленное прогрессирование соматических расстройств и нарушений ЦНС. Грубые черты лица, низкий рост, множественные дизостозы, контрактуры суставов и умственная отсталость проявляются в возрасте от 2 до 4 лет.
У некоторых пациентов на коже наблюдаются сгруппированные в очаг папулы, преимущественно в области лопаток, наружных и боковых поверхностей плеч и бедер («морская галька»), обусловленные отложением липидов и ГАГ в дерме. Характерны гипертрихоз, низкая линия роста волос на лбу, длинные густые ресницы и брови*.
P.S. * КР Союза педиатров России, Общества медицинских генетиков, «Мукополисахаридоз I», 2019 г.
У пациентов африканского и азиатского происхождения встречаются крупные монгольские пятна, которые м.б. ранним признаком болезни. Офтальмологические нарушения: пигментная дегенерация сетчатки; умеренно выраженное помутнение роговицы; у пациентов с тяжелой формой мукополисахаридоза II часто выявляется дистрофия сетчатки, приводящая к нарушению периферического и снижению сумеречного зрения; возможен отек диска зрительного нерва, обусловленный повышением ВЧД; редко встречается глаукома.
Поражение ЖКТ может привести к хронической диарее. Сообщающаяся гидроцефалия и спастическая параплегия возникают в результате утолщения мозговых оболочек. У пациентов с тяжелой формой заболевания постепенное прогрессирование многочисленных неврологических расстройств приводит к летальному исходу.
У пациентов с легкой формой заболевания отмечается почти нормальная или нормальная продолжительность жизни, минимальное поражение ЦНС и медленное прогрессирование соматических расстройств с сохранением когнитивных функций во взрослом возрасте. Имеются данные о том, что некоторые пациенты достигали возраста 65 и 87 лет и имели детей. Соматические расстройства напоминают синдром Гурлер, но в более легкой форме и с низкой скоростью прогрессирования. Рост взрослого человека может превышать 150 см. Наиболее распространенные симптомы включают поражение ДП, пороки клапанов сердца, нарушение слуха, синдром запястного канала и контрактуры суставов, которые могут привести к потере функциональности как в легкой, так и в тяжелой форме.
в) Мукополисахаридоз III типа:
1. Синдром Санфилиппо (мукополисахаридоз-III) — это гетерогенная по генетическим, но сходная по клиническим признакам группа из четырех известных типов заболевания (IIIA-IIID). Каждый тип возникает в результате дефицита разных ферментов, участвующих в разложении гепарансульфата (см. рис. 1). Гены, кодирующие эти ферменты, перечислены в табл. 2.
Синдром Санфилиппо характеризуется постепенной тяжелой дегенерацией ЦНС, сопровождающейся легкими соматическими расстройствами. Клинические признаки обычно проявляются в возрасте 2-6 лет у детей, которые ранее развивались в соответствии с нормой. К основным симптомам относят задержку когнитивного развития, гиперактивность с приступами агрессии, жесткие волосы, гирсутизм, нарушения сна и незначительную гепатоспленомегалию. Поздняя диагностика мукополисахаридоза-Ш связана с практически отсутствующими физическими отклонениями, гиперактивностью и медленно прогрессирующим неврологическим заболеванием. У большинства пациентов в возрасте 6-10 лет наблюдается резкое ухудшение неврологического статуса, которое сопровождается быстрой деградацией социальных и адаптивных навыков.
Часто встречаются серьезные поведенческие проблемы, включая нарушение сна, неконтролируемую гиперактивность, приступы гнева, деструктивное поведение и физическую агрессию. Глубокая умственная отсталость и проблемы с поведением часто возникают у пациентов с нормальным физическим развитием, что затрудняет процесс лечения.
г) Мукополисахаридоз IV типа. Синдром Моркио (мукополисахаридоз-IV) возникает в результате дефицита N-ацетилгалактозамин-6-сульфатазы (мукополисахаридоз-IVA) или β-галактозидазы (мукополисахаридоз-IVB). В обоих случаях происходит нарушение процесса разложения кератансульфата. N-ацетилгалактозамин-б-сульфатазу кодирует ген GALNS, расположенный на хромосоме в локусе 16q24.3, а β-галактозидазу — ген GLB1, расположенный на хромосоме в локусе 3р21.33. β-галактозидаза катализирует ганглиозид GM1, помимо эндогидролиза кератансульфата, поэтому в большинстве случаев мутации GLB1 приводят к возникновению генерализованного ганглиозидоза и широкого спектра нейродегенеративных расстройств, связанных со множественными дизостозами.
Мутация W273L, входящего в состав гена GLB1, в состоянии гомозиготной либо сложной гетерозиготности обычно приводит к развитию синдрома Моркио В. У пациентов с обоими типами синдрома Моркио наблюдается карликовость и дисплазия скелета, отличающаяся от дисплазии, которая встречается при др. типах мукополисахаридоза при сохранности интеллекта. Обычно мукополисахаридоз-IVA протекает в более тяжелой форме по сравнению с мукополисахаридозом-IVB, при этом рост взрослого человека составляет <125 и >150 см соответственно. Однако существует значительная вариабельность экспрессии обоих подтипов. Х-образная деформация нижних конечностей, кифоз, задержка роста с укороченным туловищем и шеей, а также переваливающаяся походка с частыми падениями являются ранними симптомами мукополисахаридоза IV.
Внескелетные проявления включают небольшое помутнение роговицы, маленькие зубы с аномально тонкой эмалью, множественный кариес, а в некоторых случаях гепатомегалию и пороки клапанов сердца. Во всех случаях наблюдается нестабильность зубовидного отростка и слабость связок, способные привести к опасной для жизни атлантоаксиальной нестабильности и смещению позвонка. Утолщение экстрадуральной ткани на передней части позвонка приводит к компрессии спинного мозга. Для таких пациентов обязательно регулярное проведение неврологических обследований и рентгенологических исследований. Операция по стабилизации верхнего ШОП (как правило, методом заднего спондилодеза) до наступления шейной миелопатии способна помочь избежать летального исхода.
д) Мукополисахаридоз VI типа. Синдром Марото-Лами (мукополисахаридоз-VI) возникает в результате мутаций гена ARSB, расположенного на хромосоме в локусе 5q11-13 и кодирующего N-ацетилгалактозамин-4-сульфатазу (арилсульфатазу В). При этом синдроме наблюдаются тяжелые или легкие соматические расстройства, как и при мукополисахаридозе-I, но при сохранности интеллекта. Соматические расстройства в тяжелой форме у пациентов с мукополисахаридозом-IV включают помутнение роговицы, грубые черты лица, контрактуры суставов, пороки клапанов сердца, сообщающуюся гидроцефалию и множественные дизостозы. При тяжелой форме заболевания рост ребенка в первые годы жизни может соответствовать норме, однако к 6-8 годам рост практически останавливается. Легкая и средняя форма синдрома Марото-Лами во многом напоминает синдром Шейе.
У пациентов с мукополисахаридозом-VI часто встречается компрессия спинного мозга из-за утолщения ТМО в верхнем цервикальном канале с последующим развитием миелопатии.
е) Мукополисахаридоз VII типа. Синдром Слая (мукополисахаридоз-VII) возникает в результате мутаций гена GUSB, расположенного на хромосоме в локусе 7q21.11. Эти мутации приводят к дефициту β-глюкуронидазы, в/клеточному накоплению фрагментов ГАГ и множественным клиническим проявлениям. В своей самой тяжелой форме синдром проявляется в виде летальной неиммунной водянки плода, которая м.б. диагностирована во время беременности с помощью УЗИ. Продолжительность жизни некоторых новорожденных с тяжелой формой заболевания может достигать нескольких месяцев, в течение которых у них проявляются симптомы лизосомных болезней накопления, включая утолщенную кожу, висцеромегалию и множественные дизостозы.
У пациентов с менее тяжелой формой мукополисахаридоза-VII в течение первых лет жизни наблюдаются симптомы, сходные с симптомами мукополисахаридоза-I, но отличающиеся более медленным прогрессированием. Также различают несколько степеней помутнения роговицы. У пациентов с симптомами, проявившимися в возрасте >4 лет, встречаются скелетные аномалии в форме множественных дизостозов, однако расстройства интеллекта и помутнение роговицы, как правило, отсутствуют. Иногда заболевание диагностируют случайно на основании результатов анализа крови, в котором видны крупные включения гранулоцитов.
ж) Мукополисахаридоз IX типа. Мукополисахаридоз-IX возникает в результате мутации гена HYAL1, расположенного на хромосоме в локусе 3р21.2-21.2 и кодирующего одну из трех гиалуронидаз. Клинические симптомы, которые наблюдались у единственной известной нам пациентки в возрасте 14 лет, включали двусторонние узелковые периартикулярные образования в мягких тканях, накопление ГАГ в лизосомах гистиоцитов, умеренную черепно-лицевую дисморфию, низкий рост, нормальное движение в суставах и сохранный интеллект. На рентгеновских снимках были обнаружены незначительные эрозии обеих вертлужных впадин.
1. Синдром «Мукополисахаридоз-плюс». У 13 детей в Северо-Восточной Сибири и у 2 детей в Турции были обнаружены грубые черты лица, органомегалия, контрактуры суставов, множественные дизостозы, когнитивная недостаточность, повышенная мукополисахаридурия и активное в/клеточное накопление гепарина сульфата. Кроме того, симптомы включали атрофию зрительного нерва, в/мозговые кальцификации, панцитопению и почечную недостаточность. Большинство пациентов скончались в течение первых двух лет жизни от кардиореспираторной недостаточности.
Активность лизосомальных ферментов у детей с синдромом «Мукополисахаридоз-плюс» соответствовала норме. Это АуР-системное поражение возникает в результате гомозиготных мутаций гена VPS33A, кодирующего белок, который принимает участие в слиянии лизосом.
з) Диагностика. При подозрении на мукополисахаридоз проводят обследование скелета. С помощью рентгеноскопии позвоночника, таза и выявляют признаки множественных дизостозов. Следующим этапом диагностики является анализ экскреции ГАГ с мочой. Полуколичественные точечные тесты на повышенную экскрецию ГАГ с мочой относятся к быстрым, недорогим и удобным методам первичной оценки, однако они могут давать как л/п, так и л/о результаты. Количественный анализ отдельных ГАГ и/или олигосахаридов с помощью масс-спектрометрических тестов выявляет типоспецифические профили в моче, сыворотке, плазме и сухих пятнах крови.
Любой человек с подозрением на мукополисахаридоз на основании клинических признаков, результатов рентгенографии или скрининговых тестов на ГАГ в моче должен иметь окончательный диагноз, установленный с помощью ферментного анализа. Сыворотка, лейкоциты или культивированные фибробласты используются в качестве источника ткани для измерения лизосомальных ферментов (см. табл. 2).
Молекулярный анализ обычно выполняется с использованием соответствующих панелей генов. Во многих случаях тип и место мутации связаны с будущим течением болезни и, т.о., имеют прогностическую ценность. Специфическая мутация также необходима, если рассматривается возможность пренатальной диагностики фетальных клеток от последующей беременности. Тестирование носительства при синдроме Хантера, связанном с Х-хромосомой, требует анализа IDS после того, как станет известна конкретная мутация или расположение хромосом в семье. Пренатальный молекулярный анализ должен быть проведен у плода мужского пола доказанной женщины-носителя гена IDS. Риск поражения такого плода составляет 50%. У плода женского пола риск невелик, но не равен нулю, поскольку искажена инактивация материнской Х-хромосомы.
Скрининг новорожденных на мукополисахаридоз возможен на основе сухих пятен крови и необходим для их раннего выявления и терапевтического вмешательства.
Муколипидозы и олигосахаридозы проявляются теми же клиническими и рентгенологическими особенностями, что и мукополисахаридозы. При этих заболеваниях показатели экскреции ГАГ с мочой не повышены. Гурлероподобные черты лица, контрактуры суставов, множественный дизостоз и повышенная экскреция ГАГ с мочой отличают мукополисахаридозы от др. нейродегенеративных и карликовых состояний.
и) Лечение. У пациентов с мукополисахаридозом II и VI отмечается значительное клиническое улучшение соматических проявлений заболевания на фоне трансплантации гематопоэтических стволовых клеток (табл. 4). Клинические эффекты включали увеличение ожидаемой продолжительности жизни с разрешением или уменьшением выраженности задержки роста, гепатоспленомегалии, скованности в суставах, внешнего вида лица, кожных изменений, СОАС, заболеваний сердца, сообщающейся гидроцефалии и тугоухости. У таких пациентов нормализуются активность фермента в сыворотке и показатели экскреции ГАГ с мочой. Вышеуказанные изменения относятся к мукополисахаридозам I-H, II и III. У пациентов с мукополисахаридозом I, которым провели трансплантацию в возрасте <9 мес, может отмечаться нормальное умственное развитие.
На фоне трансплантации в возрасте до 24 мес при исходном индексе умственного развития >70 улучшается долговременный исход. У пациентов с мукополисахаридозом, у которых на момент проведения трансплантации имеются когнитивные нарушения, данная операция не приводит к значимому улучшению нейропсихологических исходов. Тот же эффект может отмечаться при трансплантации в ранние сроки у пациентов с мукополисахаридозом II. У пациентов с мукополисахаридозом VI трансплантация приводит к стабилизации или улучшению проявлений со стороны сердца, способности поддерживать принятое положение и подвижности суставов. Трансплантация стволовых клеток не приводит к исправлению нарушений со стороны костной системы или со стороны глаз.
Заместительная ферментотерапия рекомбинантной α-L-идуронидазой одобрена к применению у пациентов с мукополисахаридозом I (см. табл. 4). На фоне применения этого фермента отмечают уменьшение выраженности органомегалии и улучшение показателей скорости роста, увеличение объема движений в суставах, сокращение количества эпизодов ночного апноэ и уменьшение экскреции ГАГ с мочой. Указанный фермент не проникает через ГЭБ и не предотвращает ухудшение нейрокогнитивной функции. Исходя из вышеописанного, заместительная ферментотерапия подходит пациентам с поражением ЦНС легкой степени или пациентам молодого возраста с целью стабилизации экстраневральных нарушений перед трансплантацией стволовых клеток.
Рекомбинантная идуронат-2-сульфатаза является ЛП выбора у пациентов с мукополисахаридозом II и применяется с целью снижения выраженности экстраневральных нарушений. На фоне заместительной ферментотерапии с рекомбинантным человеческим GALNS у пациентов с мукополисахаридозом IV отмечают повышение показателей физической выносливости, улучшение функции дыхания и увеличение активности в отношении повседневной деятельности. Сходные эффекты наблюдают у пациентов с мукополисахаридозом VI на фоне применения рекомбинантной N-ацетилгалактозамин-4-сульфатазы.
Симптоматическая терапия направлена на коррекцию осложнений со стороны дыхательной системы и ССС, ухудшения слуха, синдрома запястного канала, компрессии спинного мозга, гидроцефалии и др. проблем (табл. 5). При синдромах мукополисахаридоза, в связи с вовлечением множества систем и прогрессирующей природой, как правило, требуется комплексный подход.