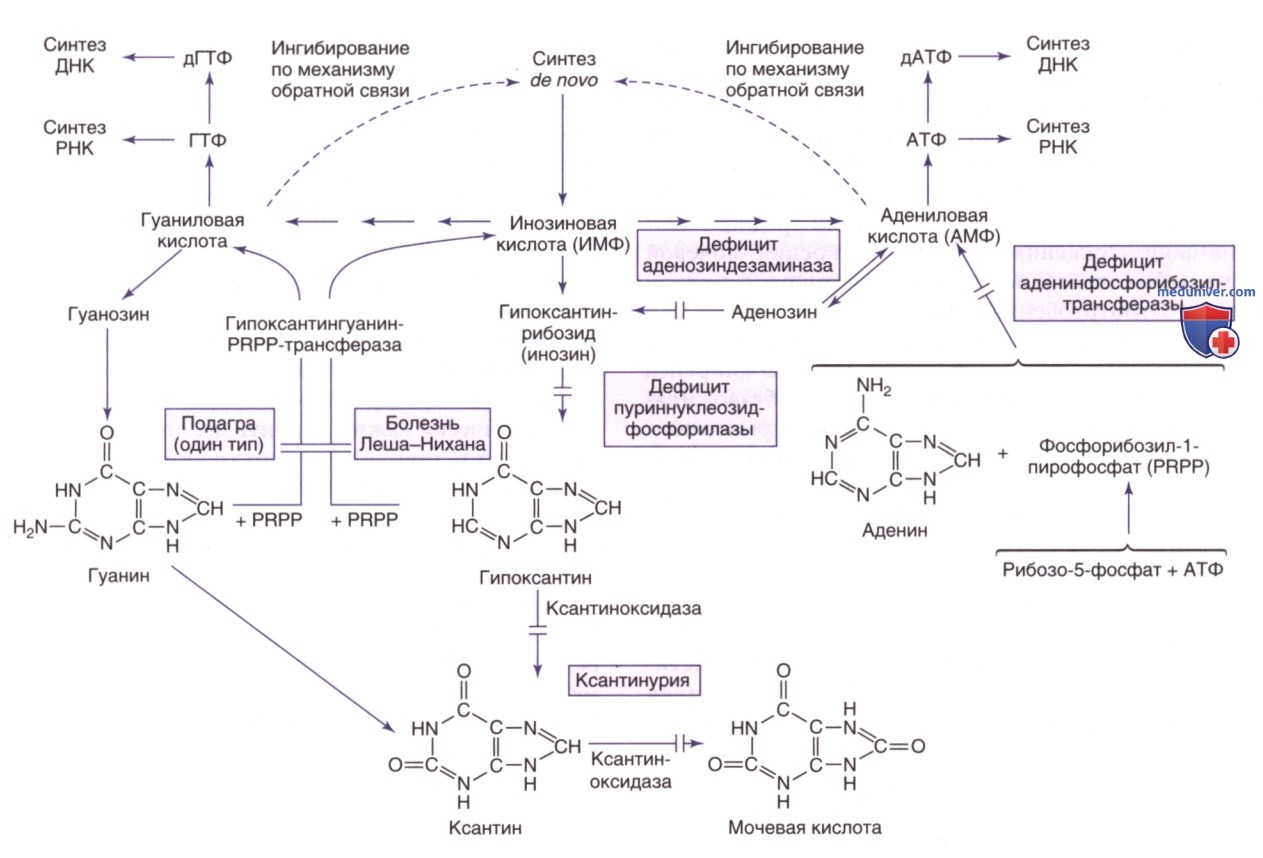
а) Повышенная активность и дефицит фосфорибозил-пирофосфат-синтетазы. Фосфорибозилпирофосфат (PRPP) — субстрат, участвующий в синтезе практически всех нуклеотидов. Он играет важную роль в регуляции синтеза пуриновых и пиримидиновых нуклеотидов de novo. Фосфорибозилпирофосфатсинтетаза (PRPS) участвует в реакции синтеза PRPP из рибозо-5-фосфата и АТФ (см. рис. 1, 2). PRPP является первым промежуточным соединением в процессе синтеза пуриновых нуклеотидов de novo, в результате которого образуется инозинмонофосфат (ИМФ), а затем АТФ и ГТФ.
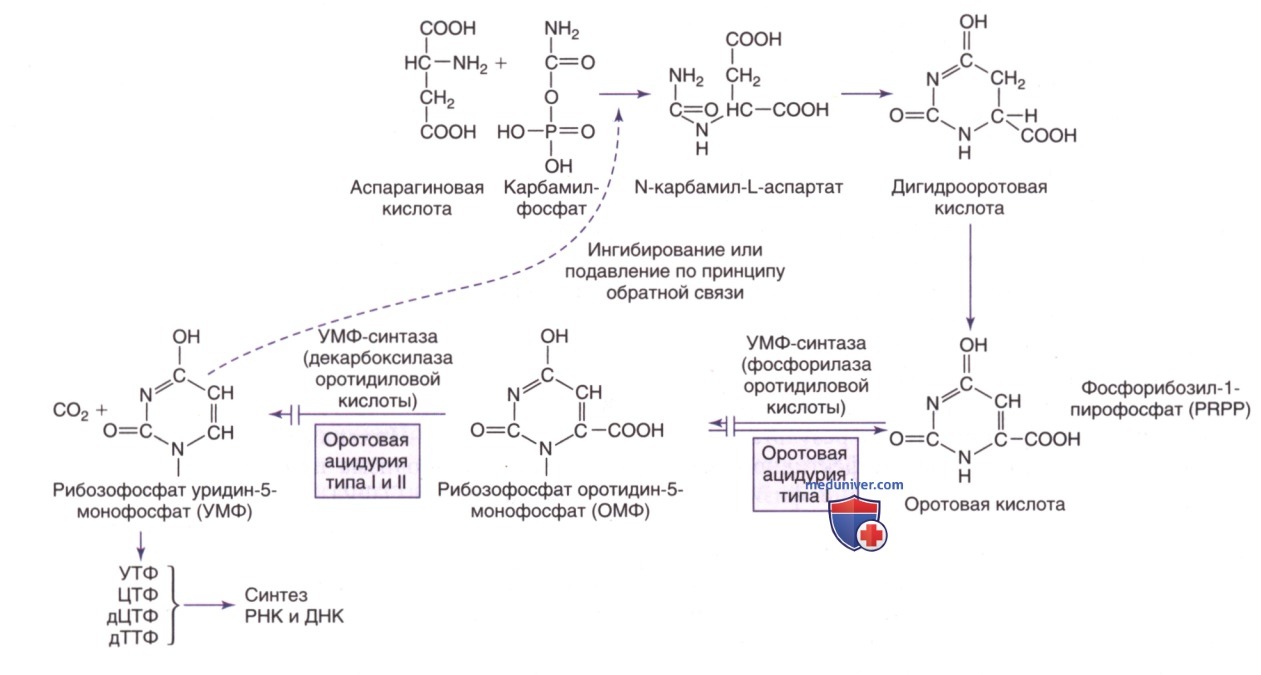
Пути биосинтеза пиримидина. PRPP — фосфорибозилпирофосфат
Генетические нарушения связаны только с изоформой PRPS-1 данного фермента. Мутации PRPS-2 не описаны. Все нарушения, связанные с PRPS, относятся к Х-сцепленным. Они делятся на суперактивность, которая может проявляться двумя фенотипами (с началом в грудном или раннем детском возрасте и более легкая форма с началом в старшем подростковом или молодом возрасте), и дефицит, проявляющийся спектром расстройств. Такие расстройства в зависимости от тяжести клинических проявлений относят к одному из трех заболеваний: синдрому Артса, Х-сцепленной болезни Шарко-Мари-Тута-5 и Х-сцепленной глухоте-2.
Повышенная активность фермента приводит к повышенному синтезу PRPP в делящихся клетках. Поскольку не происходит физиол. насыщения PRPP-аминотрансферазы, первого фермента пути синтеза пурина de novo, фосфорибозилпирофосфатом, синтез пуриновых нуклеотидов повышается, а следовательно, повышается и продукция мочевой кислоты. Повышенная активность PRPP-синтетазы — одно из немногих наследственных расстройств, при которых активность фермента повышается.
Для младенческой или ранней детской формы повышенной активности PRPS-1 характерны тяжелые неврологические последствия в сочетании с избыточной продукцией мочевой кислоты. В то же время у пациентов, у которых заболевание начинается в старшем подростковом или молодом возрасте, отклонения со стороны НС отсутствуют, однако, несмотря на это, наблюдается избыточная продукция мочевой кислоты.
Дефицит PRPS-1 приводит к истощению синтеза пуриновых нуклеотидов в тканях, зависимых от PRPS-1 (ГМ, др. нервных тканях и легких).
1. Генетика. Были клонированы и секвенированы три отдельные комплементарные ДНК, кодирующие PRPS. Две формы, PRPS-1 и PRPS-2, сцеплены с Х-хромосомой и локализуются на Xq22-q24 и Xp22.2-р.22.3 (избегают Х-инактивации), соответственно, и широко экспрессируются. Третий локус соответствует седьмой хромосоме человека и транскрибируется, по-видимому, только в яичках. Исходя из вышеописанного, дефекты PRPS-1 являются Х-сцепленными и характеризуются различной степенью тяжести клинических проявлений.
Форма повышенной активности с поздним началом связана с повышенной транскрипцией нормальной информационной РНК. Причина этого явления не установлена. Форма суперактивности с ранним началом связана с мутациями, воздействующими на аллостерическую регуляцию белка, который контролирует ингибирование неорганическими фосфатами и динуклеотидами по механизму обратной связи. В то же время указанные мутации дестабилизируют белок, что приводит к инактивации фермента в медленно реплицирующихся или нереплицирующихся клетках, напр. в нейронах и эритроцитах.
Фенотипы, сопровождающиеся дефицитом PRPS-1, напротив, связаны с мутациями, которые напрямую воздействуют на функцию фермента, обычно в месте связывания субстрата. Несмотря на то что данное нарушение является Х-сцепленным, этот вариант нужно рассматривать у всех детей или людей молодого возраста с гиперурикемией и/или гиперурикозурией и нормальной активностью ГГФРТ в лизированных эритроцитах независимо от их пола.
2. Клинические проявления. У гемизиготных мужчин с повышенной активностью фермента, проявлявшейся в грудном и раннем возрасте, наблюдаются признаки избыточной продукции мочевой кислоты, а также задержка психомоторного развития и нейросенсорная тугоухость. У таких пациентов отмечается гипотония мышц, атаксия и аутистическое поведение. Кроме того, у гетерозиготных носителей женского пола могут развиваться подагра и нарушение слуха. Тип с ранним началом обнаруживают у пациентов мужского пола, у которых отмечаются только гиперурикемия и гиперурикозурия при отсутствии неврологических симптомов.
Самая легкая форма дефицита PRPS-1 манифестирует прогрессирующей постлингвальной тугоухостью, связанной с Х-сцепленной глухотой-2 (DFN2). Более тяжелые мутации приводят к развитию фенотипа Х-сцепленной болезни Шарко-Мари-Тута-5, включающего периферическую невропатию, нарушение слуха и атрофию зрительного нерва. Большинство тяжелых мутаций PRPS-1 возникают у пациентов с синдромом Артса, который также сопровождается центральной невропатией и нарушением функций иммунной системы. У женщин заболевание, по-видимому, не проявляется, однако гемизиготные пациенты мужского пола, как правило, не живут дольше 10 лет и умирают от заболеваний легких.
Терапия SAMe продлевает жизнь, однако не оказывает видимого воздействия на симптомы неврологического дефицита, в т.ч. на тугоухость.
Механизм развития неврологических симптомов пока неизвестен. Однако можно предположить, что в нервных тканях, в т.ч. в ГМ, истощаются запасы нуклеотидов. Для дефицита PRPS-1 типичны нарушения слуха и зрения. Отсутствие этого фермента предположительно подрывает эти нервные функции с чрезвычайно высокой энергозависимостью. Кроме того, высокий уровень транскрипции PRPS-1 в легких и костном мозге позволяет предположить, что отсутствие фермента м.б. причиной рецидивирующих инфекций легких, которые характерны для синдрома Артса.
3. Лабораторные признаки. При повышенной активности PRPS-1 (как при ювенильной, так и при взрослой форме) уровень мочевой кислоты сыворотки может значительно повышаться. Кроме того, повышается уровень экскреции мочевой кислоты с мочой. При дефиците PRPS уровень мочевой кислоты не отклоняется от нормы, не снижается. Вероятно, это связано с тем, что основную часть активности по синтезу мочевой кислоты в печени и др. значимых органах обеспечивает PRPS-2. Для установления диагноза требуется определение активности PRPS-1 в эритроцитах и культуре фибробластов.
Взрослую форму суперактивности необходимо дифференцировать от частичного дефицита ГГФРТ с вовлечением пути утилизации, который также проявляется неврологическими нарушениями легкой степени или отсутствием неврологических нарушений в сочетании с гиперурикемией.
4. Лечение. Лечение дефицита PRPS, в частности синдрома Артса, заключается главным образом в экспериментальной терапии SAMe, а также в применении пищевых добавок, восполняющих истощенные запасы пуринов. Поступившие с пищей пурины не всасываются и не поступают в организм, а расщепляются в кишечнике до мочевой кислоты. У двух братьев с синдромом Артса на фоне применения SAMe (начиная с 20 мг/кг в сутки) в течение 10 лет отмечали значительное снижение частоты случаев экстренной госпитализации.
Целью терапии при суперактивности PRPS является контролирование уровня гиперурикемии с помощью аллопуринола, который ингибирует ксантиноксидазу, последний фермент пути катаболизма пуринов. Продукция мочевой кислоты снижена. Вместо мочевой кислоты синтезируется гипоксантин, который является более растворимым, и ксантин. Начальная доза аллопуринола для детей составляет 10-20 мг/кг в сутки. Дозу подбирают т.о., чтобы поддерживать нормальный уровень мочевой кислоты плазмы. Риск формирования ксантиновых камней сопоставим с риском, описанным при БЛН.
Для поддержания pH мочи на уровне 6,0-6,5 необходимо придерживаться диеты с низким содержанием пуринов (отказаться от употребления субпродуктов, сушеных бобов и сардин), употреблять большие количества жидкости и ощелачивать мочу. Указанные меры позволяют контролировать гиперурикемию и уратную нефропатию, но не влияют на неврологические симптомы. Методов лечения неврологических осложнений не разработано.
б) Дефицит аденилосукцинатлиазы (сукцинилпуринурия). Дефицит аденилосукцинатлиазы — наследственный дефицит синтеза пуринов de novo у человека. Аденилосукцинатлиаза — фермент, катализирующий два пути — синтез пуринов de novo и утилизацию пуриновых нуклеотидов. К этим путям относятся превращение сукцинил-аминоимидазол-карбокс-амидриботида (SAICAr) в аминоимидазолкарбоксамидриботид (AICAR) в пути синтеза пуриновых нуклеотидов de novo и превращение аденозилсукцината (S-АМФ) в АМФ, вторую ступень превращения инозинмонофосфата (ИМФ) в АМФ в цикле пуриновых нуклеотидов.
Дефицит аденилосукцинатлиазы приводит к накоплению в моче, СМЖ, в меньшей степени в плазме SAICAr и сукциниладенозина (S-Ado), дефосфорилированных производных SAICAr и S-АМФ соответственно.
Пути метаболизма и утилизации пуринов. PRPP — фосфорибозилпирофосфат
1. Генетика. Сукцинилпуринурия — АуР-нарушение. Ген картирован в хромосоме 22q13.1-q13.2. Выявлено ~50 генных мутаций. При проведении лабораторных исследований в моче и СМЖ обнаруживают значительное повышение уровня сукцинилпуринов, которые в норме не определяются.
2. Клинические проявления. Фатальная неонатальная форма проявляется энцефалопатией, заканчивается летальным исходом. Клинические проявления включают задержку психомоторного развития разл. степени, которая, как правило, сопровождается судорожным расстройством и аутистическим поведением (плохой зрительный контакт и стереотипные, навязчивые действия). Первыми проявлениями этого заболевания нередко становятся неонатальные судороги и тяжелая инфантильная эпилептическая энцефалопатия. В др. случаях наблюдают умственную отсталость средней или тяжелой степени, иногда сопровождающуюся задержкой роста и мышечной гипотонией.
У одной пациентки результаты тестов показали умственную отсталость легкой степени. Форма дефицита аденилосукцинатлиазы с глубокой умственной отсталостью обозначается как тип I, а вариант с умственной отсталостью легкой степени — как тип II. У др. пациентов наблюдаются клинические симптомы промежуточной степени выраженности — умеренная задержка психомоторного развития, судороги, стереотипии и возбуждение.
3. Патология. При КТ и МРТ может обнаруживаться гипотрофия или гипоплазия мозжечка, особенно червя. Предполагается, что возникновение симптомов связано скорее с нейротоксическими эффектами накопления сукцинилпуринов, чем с истощением запасов пуриновых нуклеотидов. Тяжесть фенотипических проявлений связана с соотношением S-Ado: SAICAr. Это свидетельствует о том, что SAICAr является более токсичным соединением, a S-Ado, вероятно, оказывает нейропротективное воздействие.
Лабораторная диагностика основывается на присутствии в моче и СМЖ SAICAr и S-Ado. В норме оба соединения не обнаруживаются.
4. Лечение. Эффективных методов лечения дефицита аденилосукцинатлиазы не разработано. В течение 6 мес изучали эффективность применения SAMe у младенца, у которого диагноз был установлен в раннем постнатальном периоде, однако улучшения симптомов отмечено не было. Это еще больше свидетельствует в пользу того, что заболевание связано скорее с нуклеотидной токсичностью, чем с истощением нуклеотидов. Зарегистрированы случаи пренатального установления диагноза. Младенцам и детям более старшего возраста с необъяснимой задержкой психомоторного развития или судорожным расстройством рекомендуется проводить систематическое скрининговое обследование.
в) Дефицит амино-имидазол-карбоксамид-риботид трансформилазы / инозин-монофосфат-циклогидролазы. Рибозид 5-амино-4-имизолкарбоксамид (AICA; англ. 5-amino-4-imizolecarboxamide) является дефосфорилированным продуктом аминоимидазолкарбоксамидриботида (AICAR), называемым также ZMP [5-амино-1-(5-фосфо-D-рибозил) имидазол-4-карбоксамид]. ZMP, вместе с дии трифосфатами, накапливается в эритроцитах и фиброцитах при наследственном дефиците бифункционального фермента AICAR-трансоформилазы/инозинмонофосфат(ИМФ)-циклогидролазы (ATIC), который катализирует превращение AICAR до формил-AICAR.
1. Генетика. Причиной врожденной ошибки биосинтеза пуринов заключается в мутации гена ATIC, воздействующего на активность AICAR-трансформилазы. Согласно единственному сообщению о случае, наблюдается глубокий дефицит AICAR-трансформилазы, тогда как уровень ИМФ-циклогидролазы составлял 40% от нормы.
2. Клинические особенности. Данное нарушение описано у младенцев женского пола с серьезной умственной отсталостью, эпилепсией, признаками дисморфизма (выступающие лобные бугры и метопический шов, брахицефалия, широкий рот с тонкой верхней губой, низко посаженные уши и выраженный клитор, образованный в результате сращения малых половых губ) и врожденная слепота.
3. Лабораторные признаки. Заболевание диагностируют по «+» результатам скринингового исследования мочи с использованием реакции Браттона-Маршалла с целью обнаружения AICA. Обнаруживали дефицит трансформилазы в фибробластах, что подтверждает диагноз дефицита ATIC.
4. Лечение. Эффективных методов лечения не описано.