Нейрофиброматоз (НФ) — АуД-расстройства, характеризующиеся ростом опухолей на нервах и приводящие к др. системным нарушениям. Существует три типа нейрофиброматоза: НФ1, НФ2 и шванноматоз, клинически и генетически разл. заболевания, в связи с чем должны рассматриваться как отдельные формы.
а) Клинические проявления и диагностика. НФ1 имеет частоту 1:3000 новорожденных и вызван АуД-мутациями потери функции в гене НФ1. Ок. 50% случаев заболевания носят наследственный характер, остальные 50% являются результатом спорадической мутации гена. Заболевание клинически диагностируется при наличии любых двух из следующих семи признаков.
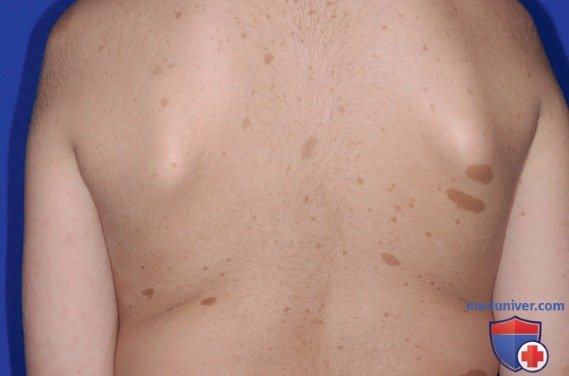
1. >6 кофейных пятен >5 мм в наибольшем диаметре у детей в препубертате и >15 мм в наибольшем диаметре у детей в постпубертате (рис. 1). Кофейные пятна являются отличительной чертой нейрофиброматоза и имеются почти у 100% пациентов. Они есть при рождении, но увеличиваются в размерах, в количестве и пигментации, особенно в первые несколько лет жизни.
Рисунок 1. Нейрофиброматоз 1-го типа. Наличие ≥6 пятен цвета кофе с молоком диаметром >0,5 см у детей и 1,5 см у подростков наводит на мысль о возможности развития нейрофиброматоза 1-го типа, хотя наличие одних только этих пятен не позволяет поставить окончательный диагноз.
Кофейные пятна рассеяны по всему телу, преимущественно на туловище и конечностях. Они не специфичны для НФ1 и могут наблюдаться при др. синдромах (табл. 2).
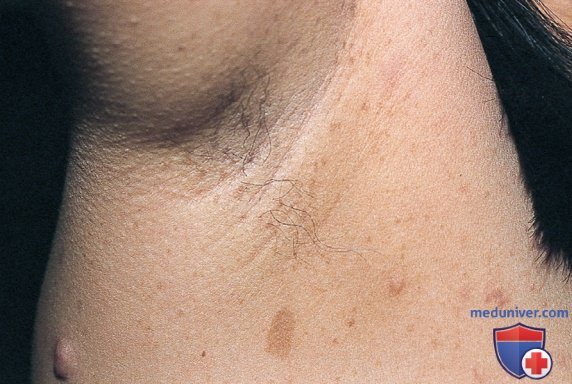
2. Подмышечные/паховые веснушки, состоящие из множества гиперпигментированных участков диаметром 2-3 мм (рис. 2). Веснушки на коже обычно появляются в 3-5 лет. Частота появления подмышечных/ паховых веснушек, как сообщается, составляет >80% к 6 годам.
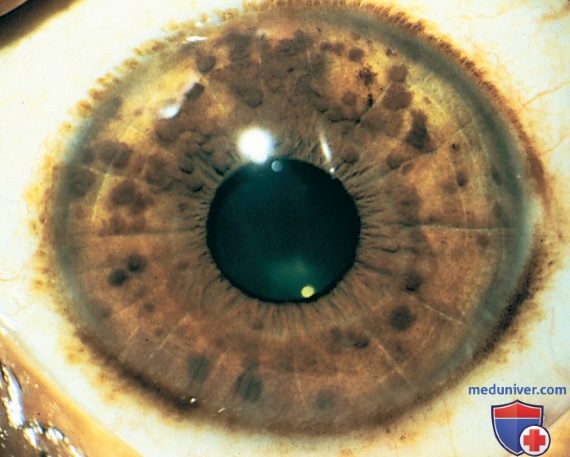
3. Два или более узелков Лиша (Lisch) на радужке, представляющие собой гамартомы, расположенные внутри радужки и лучше всего идентифицируемые при исследовании щелевой лампой (рис. 3). Они присутствуют у >74% пациентов с НФ1. Распространенность узелков Лиша увеличивается с возрастом, начиная с 5% детей <3 лет, до 42% детей 3-4 лет и практически у 100% взрослых >21 года.
Рисунок 3. Нейрофиброматоз 1-го типа. Пигментированные гамартомы радужной оболочки (узелки Лиша).
4. >2 нейрофибромы/1 плексиформная нейрофиброма. Нейрофибромы наиболее заметны на коже, но могут возникать на любом периферическом нерве, в том числе вдоль периферических нервов и кровеносных сосудов и во внутренних органах, включая ЖКТ. Их появление характерно для подросткового возраста/во время беременности, что свидетельствует о гормональном влиянии.
Обычно это небольшие, эластичные очаги с легким пурпурным обесцвечиванием вышележащей кожи. Плексиформные нейрофибромы обычно являются врожденными и возникают в результате диффузного утолщения нервных стволов и окружающих мягких тканей. Кожа, покрывающая плексиформную нейрофиброму, м.б. грубой и гиперпигментирован-ной. Плексиформные нейрофибромы могут приводить к чрезмерному росту конечности и деформации соответствующей кости.
5. Характерное поражение костей, напр. клиновидная дисплазия (которая может вызвать пульсирующий эк-зофтальм)/кортикальное истончение длинных костей с псевдоартрозом или без него (чаще всего большеберцовой кости).
6. Оптические глиомы присутствуют у 15-20% людей с НФ1; однако только 30% из них клинически значимы и требуют противоопухолевой терапии. Они являются наиболее часто наблюдаемыми опухолями ЦНС при НФ1. Из-за снижения остроты зрения детям с НФ1 рекомендуется по меньшей мере ежегодно/чаще при наличии настороженности проводить офтальмологическое обследование.
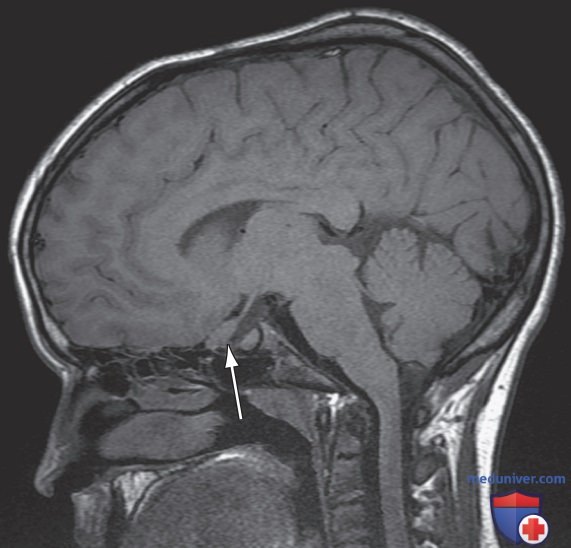
Наиболее часто симптомы развиваются в 2-6 лет; представлены изменением остроты зрения/полей зрения/бледности зрительного нерва. Глиомы в гипоталамусе могут привести к преждевременному половому созреванию. МРТ ГМ при оптической глиоме характеризуется диффузным утолщением, локализованным увеличением и наличием отчетливого очагового объемного образования, происходящего из зрительного нерва/хиазмы (рис. 4).
Рисунок 4. Оптическая глиома. Сагиттальная Т1-взвешенная магнитно-резонансная томограмма пациента с нейрофиброматозом 1-го типа демонстрирует утолщение зрительного нерва (стрелка)
7. Родственник первой степени с НФ1, диагноз которого был основан на вышеупомянутых критериях.
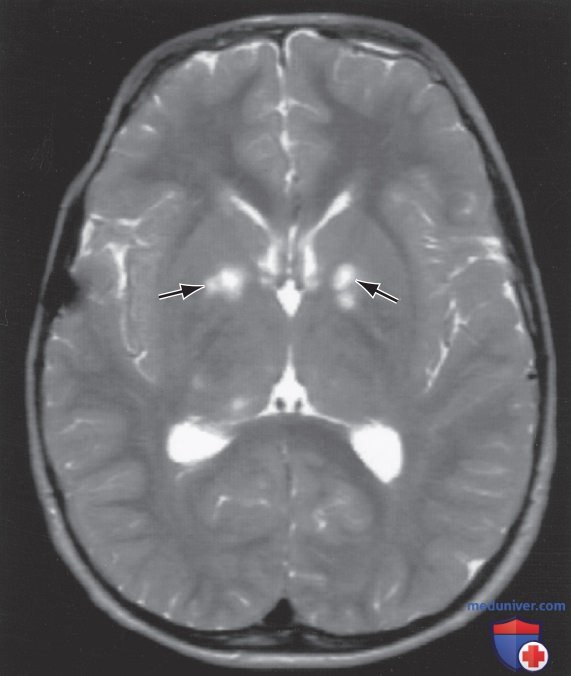
Дети с НФ1 подвержены неврологическим осложнениям. MPT-исследования отдельных детей показали аномальные гиперинтенсивные Т2-взвешенные сигналы в оптических трактах, стволе ГМ, бледном шаре, таламусе, внутренней капсуле и мозжечке (рис. 5). Эти сигналы, нечеткие яркие объекты, имеют тенденцию исчезать с возрастом; большинство из них исчезло к 30 годам.
Рисунок 5. Т2-взвешенная магнитно-резонансная томограмма пациента с нейрофиброматозом 1-го типа. Обратите внимание на зоны высокого сигнала (неопознанные яркие объекты) в базальных ганглиях (стрелки)
Неясно, что представляют собой эти очаги с патологической точки зрения, существуют разногласия относительно связи между наличием и количеством неопознанных очагов и возникновением сложностей с обучением, СДВГ, поведенческих, психосоциальных проблем и нарушений речи у детей с данной патологией. Поэтому визуализирующие исследования, такие как МРТ ГМ, должны проводиться только пациентам с клиническими симптомами.
Одним из наиболее распространенных осложнений является неспособность к обучению, имеющаяся у более половины детей с НФ1. Судороги наблюдаются у ~8% пациентов. В мозговых сосудах могут развиться аневризмы/стенозы, соответствующие синдрому мойя-мойя. Неврологические последствия этих сосудистых нарушений включают цереброваскулярные ТИА, гемипарезы и когнитивные дефекты. Преждевременное половое созревание может возникать при наличии/отсутствии поражения зрительного тракта опухолями.
ЗНО оболочки периферических нервов относятся к семейству агрессивных сарком и возникают либо de novo, либо в результате злокачественной дегенерации существующей плексиформной нейрофибромы. Пожизненный риск составляет 8-13%. Кроме того, частота феохромоцитомы, рабдомиосаркомы, лейкоза и опухоли Вильмса (Wilms) выше, чем в общей популяции. Сколиоз является распространенным осложнением, встречающимся у 10% пациентов. Пациенты с НФ1 подвержены риску развития АГ, которая м.б. изолированной/результатом стеноза почечных сосудов/ феохромоцитомы.
Мозаичный НФ1 (также называемый сегментарным НФ1) представлен поражением одного/нескольких сегментов тела, является следствием соматической (или гонадной) мутации, характерной для этой локализации. Поражения м.б. односторонними/двусторонними, асим-метричными/симметричными и ограничиваться узкой полосой/одним квадрантом. Неврологические проявления редки, но возможны.
б) Наблюдение. Из-за разнообразных и непредсказуемых осложнений, обусловленных НФ1, необходимо тщательное междисциплинарное наблюдение. Пациенты с данной патологией должны проходить регулярные клинические обследования не реже 1 p/год, обращая внимание на анамнез и выявление потенциальных осложнений, из-за которых они подвергаются повышенному риску. Наблюдение включает ежегодное офтальмологическое и неврологическое обследование, мониторинг АД и выявление сколиоза.
По мере необходимости следует проводить нейропсихологическое и интеллектуальное тестирование. Конференция по разработке консенсуса Национального института 30 рекомендовала отказаться от рутинных нейровизуализирующих исследований ГМ и зрительных путей, поскольку лечение у бессимптомных детей с НФ1 требуется редко. Однако все случаи с клиническими симптомами (напр., нарушения зрения, катаракта, повышение ВЧД) должны быть обследованы без промедления.
Селуметиниб, пероральный ингибитор МАРК-киназы 1 и 2, участвовал в предварительных испытаниях у детей с неоперабельными плексиформными нейрофибромами при НФ1, был получен частичный ответ на терапию, ЛП оказался эффективным в снижении прогрессирования опухоли.
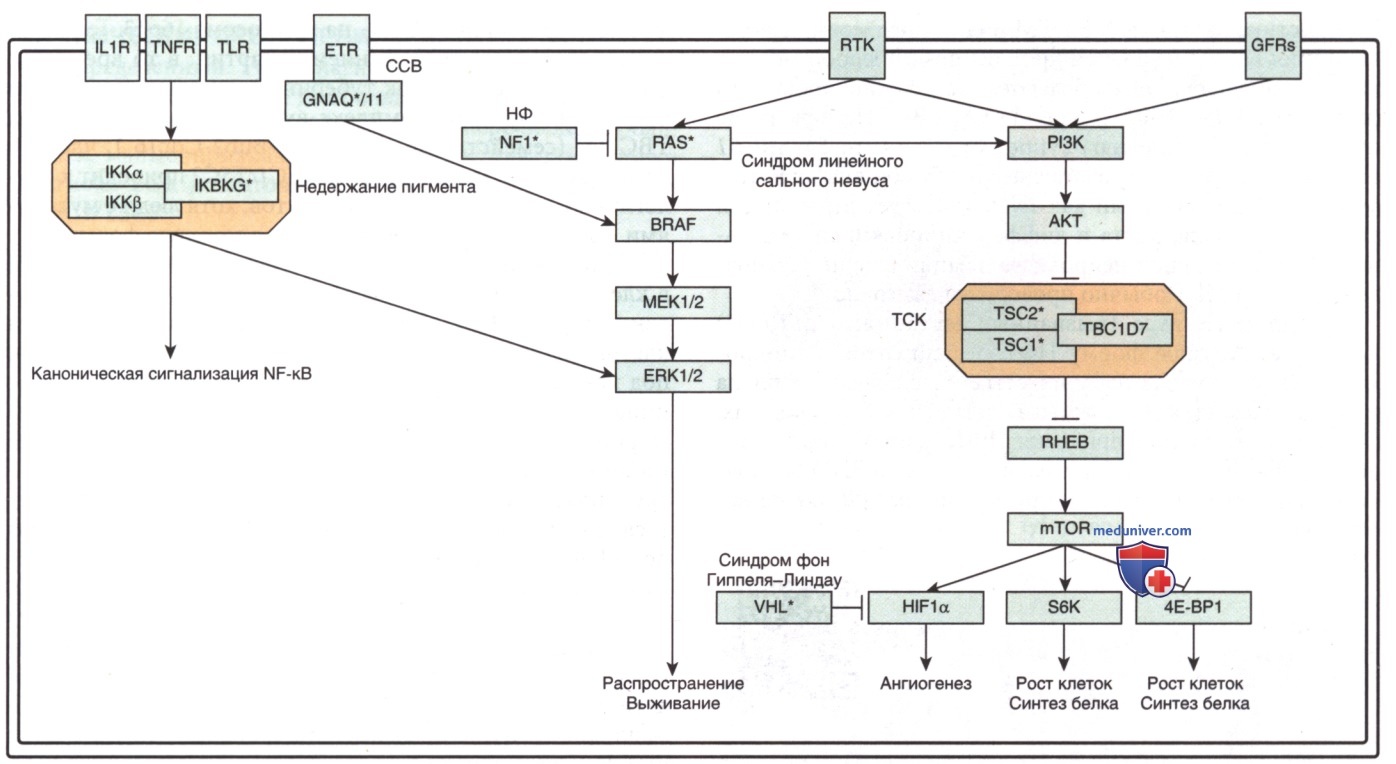
в) Генетическое консультирование. Хотя НФ1 является АуД-расстройством, более половины случаев возникают в результате спорадической мутации, представляя собой мутации de novo. Ген НФ1 в области хромосомы 17q11.2 кодирует белок, известный как нейрофибромин. Он действует как ингибитор онкогена Ras (рис. 6). Диагноз НФ1 основывается на клинических особенностях. Однако молекулярное тестирование мутаций гена НФ1 доступно и м.б. использовано в ряде случаев.
Рисунок 6. Схематическое представление клеточных путей, затронутых мутациями в генах, вызывающих нейрокожные нарушения, напр. нейрофиброматоз 1-го типа, туберозный склерозный комплекс и синдром Стерджа-Вебера. Звездочки обозначают гены синдромов, обсуждаемых в отдельных статьях на сайте (просим Вас пользоваться формой поиска по сайту выше)
Генетическое тестирование необходимо пациентам, соответствующим только одному критерию клинического диагноза, пациентам с тяжелой формой заболевания и при проведении пренатальной/предимплантационной диагностики.
НФ2 является менее распространенным заболеванием, чем НФ1; передается АуД-путем, с частотой 1:25 000 родов. Клинические диагностические критерии установлены консенсусной конференцией Национальных институтов ЗО США и модифицированы в манчестерские и базовые критерии. Диагноз м.б. подтвержден генетическим анализом крови/идентичной мутацией в двух отдельных опухолях от одного и того же индивидуума. Как правило, НФ2 диагностируется при наличии одного из следующих четырех признаков:
1) билатеральные вестибулярные шванномы;
2) родитель, брат, ребенок с НФ2 и односторонней вестибулярной шванномой/любыми двумя из следующих: менингиомой, шванномой, глиомой, нейрофибромой, задними субкапсулярными линзовидными помутнениями;
3) односторонняя вестибулярная шваннома и любые две из следующих: менингиома, шваннома, глиома, нейрофиброма, заднее субкапсулярное линзовидное помутнение;
4) множественные менингиомы (>2) и односторонняя вестибулярная шваннома/любые две из следующих: шваннома, глиома, нейрофиброма, катаракта.
В детском возрасте могут появиться шум в ушах, потеря слуха, головная боль и неустойчивость при ходьбе, хотя признаки поражения мозжечкового угла чаще возникают во втором и третьем десятилетиях жизни. Кофейные пятна и плексиформные шванномы кожи отмечаются уже в педиатрической возрастной группе. С помощью щелевой лампы выявляются помутнения хрусталика у ~50% пациентов с НФ2. Ген НФ2 (который кодирует белок, известный как Мерлин/шванномин) расположен на хромосоме 22q1.11. В табл. 3 указана частота развития патологии при НФ2.
Офтальмологическое обследование, МРТ ГМ и спинного мозга, аудиометрия и вызванные стволовые потенциалы — важные компоненты постоянного наблюдения пациентов с НФ2. Вследствие частого развития множественных сопутствующих опухолей в/черепная патология лечится консервативно с целью сохранения слуха и максимального повышения качества жизни.
Шванноматоз — форма НФ, клинически отличная от НФ1 и НФ2, характеризуется множественными шванномами при отсутствии двусторонних вестибулярных шванном. Хотя общая заболеваемость значительно ниже — 0,47:1 000 000 человек, считается, что лица со шванноматозом составляют 2-10% всех лиц, подвергшихся хирургической резекции по поводу шванномы. Подсчитано, что по меньшей мере 20% из них носят семейный характер.
Диагноз подразумевается у человека с множественными шванномами, особенно при наличии больного члена семьи. Диагностика включает МРТ ГМ и спинного мозга для исключения вестибулярных и др. шванном.
В любой момент наблюдения первичное обследование должно проводить различие между НФ2 и шванноматозом. Исследование привело к открытию гена-супрессора опухоли SMARCB1 как основного предрасполагающего гена при шванноматозе. SMARCB1, известный как INI1, участвует в регуляции клеточного цикла, роста и дифференцировки. Оптимальная частота обзорной нейровизуализации еще не установлены, однако МРТ обычно проводится ежегодно.
Синдром Легиуса (вызванный мутациями SPRED1) напоминает легкую форму НФ1. У пациентов с синдромом Легиуса отмечаются множественные кофейные пятна и макроцефалия, как с кожными складками, так и без них. Однако др. типичные признаки НФ1, напр. узелки Лиша, нейрофибромы, глиомы зрительного нерва и ЗНО оболочки периферических нервов, при мутациях SPRED1 не наблюдаются.