Заболевания печени вызывает широкий спектр митохондриальных нарушений. В гепатоцитах наблюдается высокая плотность митохондрий, потому что печень с ее биосинтетическими и детоксицирующими функциями сильно зависит от АТФ. Изменения функций митохондрий могут привести к нарушению окислительного фосфорилирования, увеличению выработки активных форм кислорода, нарушению др. метаболических путей и активации механизмов клеточной гибели.
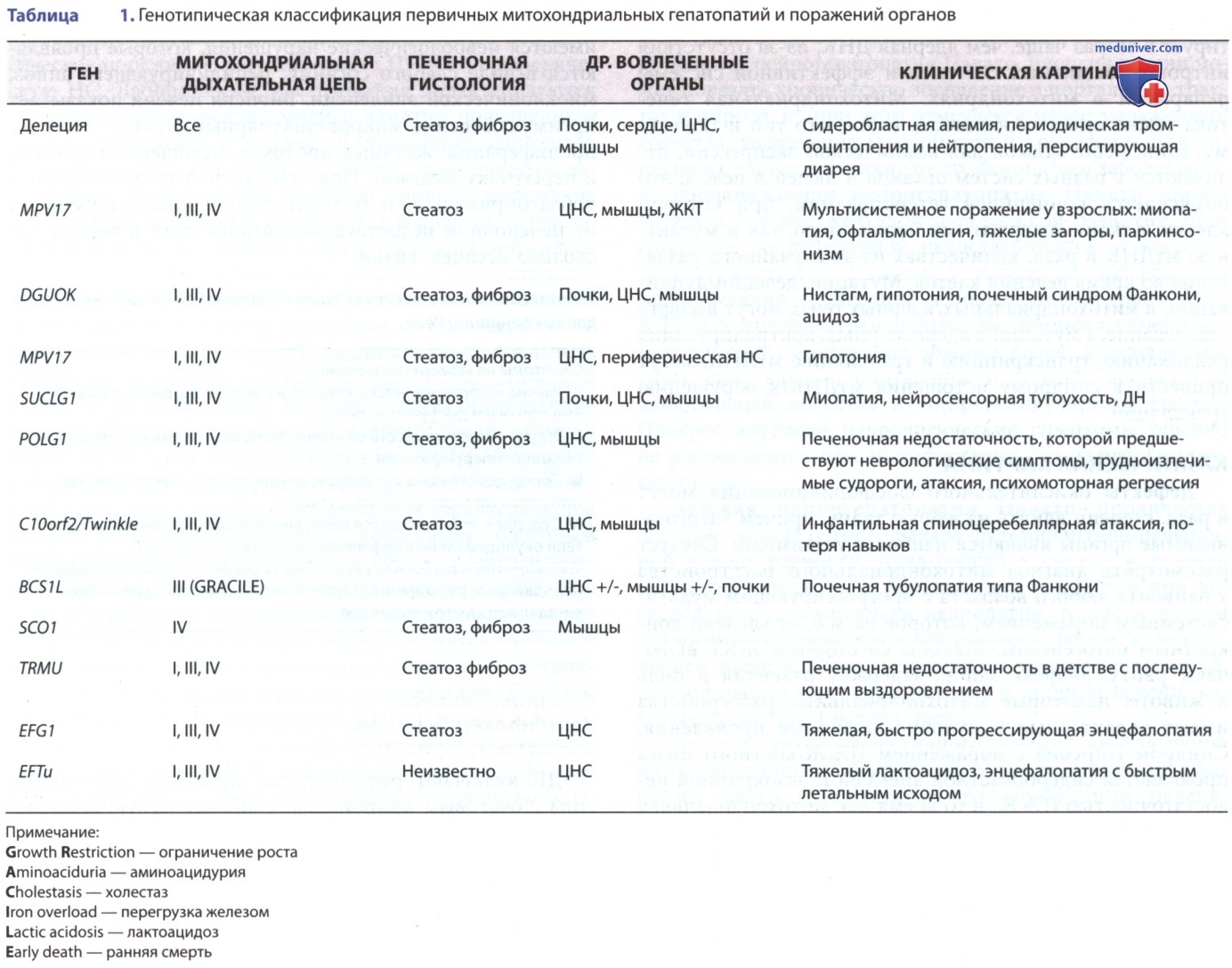
Митохондриальные нарушения можно разделить на первичные, при которых митохондриальный дефект является основной причиной нарушения, и вторичные, при которых функция митохондрий нарушается в результате экзогенного повреждения/генетической мутации, ввиду чего нарушается синтез немитохондриальных белков. Первичные митохондриальные нарушения м.б. вызваны мутациями, влияющими на митохондриальную ДНК (мтДНК)/ядерными генами, которые кодируют митохондриальные белки/кофакторы (табл. 1).
Можно отметить конкретные закономерности (табл. 2). Вторичные митохондриальные нарушения включают заболевания с неопределенной этиологией, такие как синдром Рейе, расстройства, вызванные эндогенными/экзогенными токсинами, ЛП/металлами и др. состояния, при которых окислительное повреждение митохондрий м.б. вовлечено в патогенез повреждения печени.
а) Эпидемиология. Нарушения митохондриальной дыхательной цепи всех типов поражают 1:20 000 детей <16 лет; поражение печени зарегистрировано у 10-20% пациентов с дефектом дыхательной цепи. Первичные митохондриальные нарушения, включая синдромы истощения мтДНК, встречаются у 1:5000 живорожденных и являются одной из известных причин острой печеночной недостаточности у детей <2 лет.
Идентифицировано >200 патогенных точечных мутаций, делеций, вставок и перегруппировок, которые включают мтДНК и ядерную ДНК и кодируют митохондриальные белки. Митохондриальная генетика уникальна тем, что митохондрии способны самостоятельно реплицировать, транскрибировать и переводить свою мтДНК.
Типичный гепатоцит содержит 1000 копий мтДНК. Окислительное фосфорилирование (процесс получения АТФ) происходит в дыхательной цепи, расположенной во внутренней мембране митохондрий, и разделено на пять мультиэнзимных комплексов: восстановленная никотинамидадениндинуклеотидная коэнзим Q редуктаза (комплекс I), сукцинат-коэнзим Q редуктаза (комплекс II), восстановленная коэнзим Q-цитохром-с редуктаза (комплекс III), цитохром-с оксидаза (комплекс IV) и аденозинтрифосфатсинтаза (комплекс V).
Пептидные компоненты дыхательной цепи кодируются как ядерными генами, так и генами мтДНК, поэтому мутации в любом геноме могут привести к нарушениям окислительного фосфорилирования. Тринадцать основных полипептидов синтезируются из небольшой 16,5 кб круговой двухцепочечной мтДНК. мтДНК кодирует 24 передаточные РНК, необходимые для в/митохондриального синтеза белка, в то время как ядерные гены кодируют >70 субъединиц дыхательной цепи и множество ферментов и кофакторов, необходимых для поддержания мтДНК, включая ДНК-полимеразу-γ (POLG), тимидинкиназу 2 и дезоксигуанозинкиназу.
Выявление митохондриальных нарушений затруднено, и эпидемиологические исследования сопряжены с техническими трудностями при сборе и обработке образцов тканей, необходимых для постановки точного диагноза, вариабельностью клинической картины и тем фактом, что большинство нарушений имеет вариабельную пенетрантность и наследуется от матери. МтДНК мутирует в 10 раз чаще, чем ядерная ДНК, из-за отсутствия интронов, защитных гистонов и эффективной системы репарации в митохондриях.
Митохондриальная генетика также имеет особенность в том, что тип и тяжесть мутации, необходимой для клинической экспрессии, отличаются у разных систем органов и людей в целом; это объясняется концепцией гетероплазмии, при которой клетки и ткани содержат как нормальную, так и мутантную мтДНК в разл. количествах из-за случайного разделения во время деления клеток. Мутации, делеции/дупли-кации, в митохондриальных/ядерных генах могут вызвать заболевание, а мутации в ядерных генах, контролирующих репликацию, транскрипцию и трансляцию мтДНК, могут привести к синдрому истощения мтДНК/к нарушению трансляции.
б) Клиническая картина. Дефекты окислительного фосфорилирования могут в разл. степени влиять на любую ткань, причем энергозависимые органы являются наиболее уязвимыми. Следует рассмотреть диагноз митохондриального расстройства у пациента любого возраста с прогрессирующим мультисистемным поражением, которое не м.б. объяснено конкретным патогенезом. Жалобы со стороны ЖКТ включают рвоту, диарею, запор, задержку развития и боль в животе; некоторые митохондриальные расстройства имеют характерные желудочно-кишечные проявления.
Синдром Пирсона с поражением ПЖЖ/костного мозга проявляется сидеробластной анемией и экзокринной недостаточностью ПЖЖ, в то время как митохондриальная нейрогастрокишечная энцефаломиопатия проявляется хронической кишечной псевдообструкцией и кахексией. Печеночные проявления варьируют от хронического холестаза, гепатомегалии, цирроза и стеатоза до молниеносной печеночной недостаточности и смерти. У пациентов с определенными митохондриальными заболеваниями м.б. нормальный/минимально повышенный уровень лактата даже в условиях метаболического криза.
Молярное соотношение лактат/пируват было предложено в качестве теста-скрининга для выявления митохондриальных нарушений, поскольку оно отражает равновесие между продуктом и субстратом реакции, катализируемой ЛДГ. Считается, что соотношение лактат/пируват >25 в значительной степени указывает на дисфункцию дыхательной цепи; однако повышенный уровень соотношения м.б. при вторичной митохондриальной дисфункции, возникающей в результате тяжелого заболевания печени.
в) Первичные митохондриальные гепатопатии:
1. Неонатальная печеночная недостаточность. Распространенным проявлением дефектов дыхательной цепи является тяжелая печеночная недостаточность, проявляющаяся желтухой, гипогликемией, коагулопатией, нарушением функции почек и гипераммонемией, начинающаяся в первые несколько недель/месяцев жизни. Дефицит цитохром-с-оксидазы (комплекса IV) является наиболее распространенным дефицитом у этих детей, хотя нарушения со стороны комплексов I и III и синдром истощения мтДНК также имеют место (см. табл. 1 и 3).
Ключевые биохимические особенности включают заметно повышенную концентрацию лактата в плазме, повышенное молярное соотношение лактат/пируват (>25) и повышенное соотношение β-гидроксибутирата к ацетоацетату (>4,0). Симптомы неспецифичны, включают вялость и рвоту. У большинства пациентов дополнительно имеются неврологические нарушения, которые проявляются в виде слабого сосания, рецидивирующего апноэ, миоклонической эпилепсии. Биопсия печени показывает преимущественно микровезикулярный стеатоз, холестаз, пролиферацию желчных протоков, истощение гликогена и перегрузку железом. При стандартной терапии прогноз неблагоприятный, и большинство пациентов умирают от печеночной недостаточности/инфекции в первые несколько месяцев жизни.
2. Синдром Альперса (синдром Альперса-Хаттенлохера или гепатопатическая полиодистрофия Альперса). ДК включают рефрактерные припадки смешанного типа с очаговым компонентом; эпизодическую психомоторную регрессию, вызванную интеркуррентными инфекциями, и гепатопатию с острой печеночной недостаточностью или без нее. Синдром Альперса проявляется с младенчества до 8 лет судорогами, гипотонией, трудностями с кормлением, психомоторной регрессией и атаксией.
У пациентов развиваются гепатомегалия и желтуха, а прогрессирование печеночной недостаточности протекает медленней, чем у пациентов с дефицитом цитохрома-с-оксидазы. Подтверждают диагноз повышенные уровни лактата и пирувата в крови/СМЖ, в дополнение к характерным данным ЭЭГ (высокоамплитудная медленная активность с полиспайками), асимметричным аномальным зрительно вызванным реакциям и областям низкой плотности/атрофии в затылочных/височных долях на КТ ГМ. У некоторых пациентов был обнаружен дефицит комплекса I в митохондриях печени/мышц. Тип наследования заболевания — АуР; во многих семьях с синдромом Альперса были идентифицированы мутации в каталитической субъединице ядерного гена POLG мтДНК, что привело к появлению молекулярной диагностики синдрома Альперса. Пациенты с мутациями POLG восприимчивы к вальпроат-индуцированной дисфункции печени.
3. Синдром истощения митохондриальной дезоксирибонуклеиновой кислоты. Синдром истощения мтДНК характеризуется тканеспецифическим снижением числа копий мтДНК, что приводит к дефициту комплексов I, III и IV. Синдром истощения мтДНК проявляется фенотипической гетерогенностью; мультисистемные и локализованные формы заболевания включают миопатические, гепатоцеребральные и гепатопатические проявления. У младенцев гепатоцеребральная форма чаще диагностируется в неонатальном периоде. Первые симптомы — метаболические, быстро прогрессирующие до печеночной недостаточности с гипогликемией и рвотой. За этой стадией следует неврологическое поражение, затрагивающее ЦНС и периферическую НС. Лабораторные исследования характеризуются лактоацидозом, гипогликемией и заметно повышенным содержанием АФП в плазме.
У некоторых пациентов была обнаружена перегрузка железом с повышенным насыщением трансферрина, высоким уровнем ферритина и накоплением железа в гепатоцитах и клетках Купфера. Смерть обычно наступает к 1-му году жизни. Сообщалось о спонтанном выздоровлении пациента с гепатопатией. Наследование по АуР-типу и мутации в гене DGUOK были выявлены у многих пациентов с гепатоцеребральной формой заболевания. Тимидинкиназа-2 вовлечена в развитие миопатической формы; при гепатопатической форме не было выявлено ни одного известного генетического дефекта.
Множество др. ядерных генов, включая POLG, MPV17, ген геликазы мерцания и SUCLG1, вызывают развитие гепатоцеребральной формы. Биопсия печени пациентов с данным заболеванием характеризуется микровезикулярным стеатозом, холестазом, очаговым цитоплазматическим билиарным некрозом и цитосидерозом в гепатоцитах и синусоидальных клетках. Характерны ультраструктурные изменения с онкоцитарной трансформацией митохондрий, которая характеризуется наличием митохондрий с редкими кристаллами, зернистым матриксом и плотными/везикулярными включениями.
Если нативный комплекс II, кодируемый ДНК, в норме, а активность др. комплексов снижена, необходимо исследовать количество копий мтДНК для диагностики синдрома истощения мтДНК. Диагноз устанавливается при низком соотношении мтДНК (<10%) к ядерной ДНК в пораженных тканях и/или посредством генетического тестирования. Важно отметить, что последовательность митохондриального генома в норме.
4. Нейрогепатопатия Навахо. Нейрогепатопатия Навахо — АуР-наследуемая сенсомоторная нейропатия с прогрессирующим заболеванием печени, встречающаяся только у индейцев Навахо на юго-западе США. Заболеваемость 1:1600 живорождений. ДК включают сенсорную нейропатию, двигательную нейропатию, потерю чувствительности роговицы, заболевание печени, метаболические/инфекционные осложнения, включая задержку развития, низкий рост, задержку полового созревания, системную инфекцию; и доказательства демиелинизации ЦНС при рентгенографии и биопсии периферических нервов.
В патогенезе нейрогепатопатии Навахо отмечается мутация гена MPV17. Интересно, что тот же самый ген вовлечен в развитие синдрома истощения мтДНК (см. ранее), что доказывает, что нейрогепатопатия Навахо м.б. специфическим типом синдрома истощения мтДНК, встречающимся только у индейцев Навахо. Данное заболевание делится на три фенотипических варианта в зависимости от возраста появления и клинической картины.
- Классическая нейрогепатопатия Навахо проявляется в младенчестве тяжелым прогрессирующим неврологическим ухудшением в виде слабости, гипотонии, потери чувствительности с сопутствующими повреждением конечностей, изъязвлениями роговицы и замедленным ростом. Заболевание печени, встречающееся у большинства пациентов, является вторичным и варьирует от бессимптомного повышения функциональных показателей печени до эпизодов, подобных синдрому Рейе, гепатоцеллюлярной карциномы и цирроза. Уровни γ-глутамилтранспептидазы, как правило, выше, чем при др. формах нейрогепатопатии Навахо. Биопсия печени может показать хроническое воспаление в портальных трактах и цирроз, но при этом отмечается менее выраженный холестаз, увеличение гепатоцитов и трансформация гигантских клеток в отличие от др. форм этого заболевания.
- Инфантильная нейрогепатопатия Навахо проявляется в 1-6 мес желтухой и задержкой развития, прогрессируя до печеночной недостаточности, и заканчивается смертью к 2 годам. У пациентов наблюдается гепатомегалия с умеренным повышением уровня ACT, АЛТ и у-глутамилтранспептидазы. Биопсия печени демонстрирует образование ложных долек, многоядерные гигантские клетки, портальное и долевое воспаление, канальцевый холестаз и микровезикулярный стеатоз. Прогрессирующие неврологические симптомы обычно не распознаются при манифестации, но идентифицируются позже.
- Детская нейрогепатопатия Навахо проявляется в 1-5 лет в виде острого начала фульминантной печеночной недостаточности, приводящей к смерти в течение нескольких месяцев. У большинства пациентов при исследовании есть признаки невропатии. Биопсия печени аналогична таковой при инфантильной форме, за исключением значительного увеличения и некроза гепатоцитов, пролиферации желчных протоков и цирроза печени, которые также могут встречаться.
Не существует эффективного лечения ни для одной из форм данного заболевания, а неврологические симптомы часто препятствуют трансплантации печени. Идентичная мутация MPV17 наблюдается у пациентов как с инфантильной, так и с классической формами нейрогепатопатии Навахо, что подчеркивает клиническую гетерогенность заболевания.
5. Синдром Пирсона. Синдром Пирсона с поражением ПЖЖ/костного мозга развивается у новорожденных с тяжелой макроцитарной анемией, вариабельной нейтропенией, тромбоцитопенией, а также кольцевыми сидеробластами в костном мозге. В раннем детстве развиваются диарея и мальабсорбция жиров как следствие обширного фиброза ПЖЖ, ацинарной атрофии и частичной атрофии ворсинок тонкой кишки. Поражение печени проявляется гепатомегалией, стеатозом и циррозом. Сообщалось о печеночной недостаточности и смерти в возрасте <4 лет. Др. особенности синдрома включают поражение почечных канальцев, фотосенсибилизацию, СД, водянку плода и поздние проявления, среди которых нарушения зрения, тремор, атаксия, слабость проксимальных мышц, внешняя офтальмоплегия и пигментная ретинопатия.
Обнаружение метилглютаконовой ацидурии — диагностический маркер заболевания. У большинства пациентов регистрируются крупные делеции в мтДНК, приводящие к дефициту комплексов I и III. Делеции в мтДНК м.б. обнаружены в культивируемых фибробластах пациентов, а также в лимфоцитах периферической крови.
6. Синдром атрофии ворсинок. У детей с этим заболеванием на первом году жизни наблюдается тяжелая анорексия, рвота, хроническая диарея и атрофия ворсинок. Поражение печени включает умеренное повышение уровня аминотрансфераз, гепатомегалию и стеатоз. Лактоацидоз усугубляется в/в-инфузиями р-ров с высоким содержанием декстрозы/энтеральным питанием. К 5 годам облегчаются симптомы диареи и нормализуются результаты биопсии кишечника. Впоследствии у пациентов развивается пигментный ретинит, мозжечковая атаксия, нейросенсорная глухота и слабость проксимальных мышц, что в конечном итоге приводит к смерти в конце 1-го десятилетия жизни. Заболевание связано с дефектом перестройки мтДНК. В мышцах пациентов был обнаружен дефицит комплекса III.
7. Синдром GRACILE. Аббревиатура GRACILE обобщает наиболее важные клинические особенности, а именно задержку роста плода (низкая МТР ок. -4SD), аминоацидурию (вызванную тубулопатией типа Фанкони), холестаз (со стеатозом и циррозом печени), перегрузку железом, тяжелый лактоацидоз и раннюю смерть. Синдром связан с мутациями комплекса III фактора сборки BCS1L. Гистология печени обнаруживает микровезикулярный стеатоз и холестаз с обильным накоплением железа в гепатоцитах и клетках Купфера. Содержание железа в печени незначительно снижается с возрастом, что сопровождается увеличением фиброза и цирроза печени. Отмечаются аномальные уровни аминотрансфераз и свертывания крови, но причина смерти, по-видимому, больше связана с истощением энергии, чем с печеночной недостаточностью. Ок. половины этих пациентов умирают в первые 2 нед жизни.
8. Мутации в генах фактора ядерной трансляции и удлинения. Мутации в генах фактора ядерной трансляции (TRMU; митохондриальная тРНК-специфическая 2-тиоуридилаза 1) ферментных комплексов дыхательной цепи были идентифицированы как этиология острой печеночной недостаточности, проявляющейся в возрасте 1 день-6 мес. Дефицит дыхательной цепи аналогичен тому, что наблюдается при синдроме истощения мтДНК, где активность нативного комплекса II, кодируемого ДНК, в норме, в то время как активность комплексов I, III и IV снижена. Мутация фактора удлинения EFG1 [гена GFM1 (G фактор элонгации митохондрий 1)] вызывает ограничение роста плода, лактоацидоз, дисфункцию печени, которая прогрессирует в печеночную недостаточность и смерть. Мутация в факторе удлинения EFTu проявляется в виде тяжелого лактоацидоза и летальной энцефалопатии с легким поражением печени.
9. Вторичные митохондриальные гепатопатии. Вторичные митохондриальные гепатопатии вызываются воздействием гепатотоксического металла, ЛП, токсина, эндогенного метаболита. В прошлом наиболее распространенной вторичной митохондриальной гепатопатией был синдром Рейе, распространенность которого достигла пика в 1970-х гг. и имела смертность >40%. Хотя смертность не изменилась, распространенность снизилась с >500 случаев в 1980 гг. до ~35 случаев в год в настоящий момент.
Снижение зарегистрированной заболеваемости синдромом Рейе м.б. частично связано с более точной современной диагностикой инфекционных, метаболических и токсических заболеваний, что позволяет снизить процент идиопатических/истинных случаев синдрома Рейе. Синдром Рейе обнаруживается у генетически восприимчивого человека в результате взаимодействия вирусной инфекции (гриппа, ветряной оспы) и применения салицилатов и/или противорвотных ЛС.
Клинически он характеризуется предшествующим вирусным заболеванием, которое, по-видимому, проходит, и острым началом рвоты и энцефалопатии (см. табл. 3). Неврологические симптомы могут быстро прогрессировать до судорог, комы и смерти. Дисфункция печени неизменно возникает при развитии рвоты, сопровождающаяся коагулопатией и повышением сывороточных уровней ACT, АЛТ и аммиака. Важно отметить, что желтуха отсутствует и уровень билирубина в сыворотке крови остается в норме. Биопсия печени показывает микровезикулярный стеатоз без признаков воспаления/некроза печени. Смерть обычно является следствием повышенного ВЧД и развития мозговой грыжи. У выживших пациентов функция печени полностью восстанавливается, но им необходимо провести обследование на предмет дефектов окисления и переноса жирных кислот (табл. 4).
Приобретенные нарушения функции митохондрий м.б. вызваны несколькими ЛС и токсинами, включая вальпроевую кислоту, цианид, амиодарон, хлорамфеникол, железо, рвотный токсин Bacillus cereus и аналоги нуклеозидов. Вальпроевая кислота — жирная кислота с разветвленной цепью, которая может метаболизироваться в митохондриальный токсин, а именно 4-энвальпроевую кислоту. Дети с серьезными дефектами дыхательной цепи, по-видимому, более чувствительны к токсическому воздействию этого ЛП, и, как сообщается, вальпроевая кислота вызывает печеночную недостаточность у пациентов с синдромом Альперса и дефицитом цитохром-с-оксидазы.
Аналоги нуклеозидов ингибируют непосредственно комплексы дыхательных цепей митохондрий. Ингибиторы обратной транскриптазы зидовудин, диданозин, ставудин и залцитабин, используемые для лечения ВИЧ-инфицированных пациентов, ингибируют POLG и могут блокировать удлинение мтДНК, приводящее к ее истощению. Др. состояния, которые могут привести к митохондриальному окислительному стрессу, включают холестаз, неалкогольный стеатогепатит, дефицит α1-антитрипсина и болезнь Вильсона.
г) Диагностика. Скрининговые тесты включают общие биохимические тесты (комплексный метаболический профиль, МНО, АФП, КФК, фосфор, ОАК, аммиак, лактат, пируват, кетоновые тела в сыворотке крови: количественные 3-гидроксибутират и ацетоацетат, общее количество свободных жирных кислот, профиль ацилкарнитина; свободный и общий карнитин в сыворотке крови, органические кислоты мочи и аминокислоты сыворотки) (см. табл. 5). Эти результаты будут служить основой для последующего подтверждающего тестирования и установления молекулярного диагноза. Генотипирование, включая скрининг одного гена/панели на распространенные митохондриальные заболевания, широко используется в клинической практике. Также полезно секвенирование всего экзома/генома, которое заменяет тестирование одного гена/панели генов.
Однако идентификация нескольких вариантов генов неопределенной значимости потребует детального клинического и биохимического подтверждения. Ткани (биопсия печени, фибробласты кожи и биопсия мышц) могут потребоваться для постановки конкретного биохимического диагноза.
д) Лечение митохондриальных гепатопатий. Для большинства пациентов с митохондриальными гепатопатиями не существует эффективной терапии; неврологическое поражение часто исключает ортотопическую трансплантацию печени. Пациенты с митохондриальными нарушениями подвержены риску обострения основного метаболического заболевания после трансплантации, особенно пациенты с заболеваниями, связанными с POLG. Было предложено несколько комбинаций терапевтических ЛП, включая антиоксиданты, витамины, кофакторы и акцепторы электронов, но для оценки их эффективности не было проведено ни одного РКП.
Лечение представляет собой поддерживающую терапию и включает в/в-инъекции натрия гидрокарбаната («Натрия бикарбоната») при остром метаболическом ацидозе, переливание крови при анемии и тромбоцитопении и прием ферментов ПЖЖ при недостаточности ее экзогенной функции. Важно прекратить прием/избегать ЛС, которые могут усугубить гепатопатию, включая вальпроевую кислоту («Вальпроат натрия»), тетрациклин, макролидные АБ, азатиоприн, хлорамфеникол, хинолоны и линезолид. Следует избегать применение натрия хлорида раствора сложного [калия хлорида + кальция хлорида + натрия хлорида] («Рингера»), содержащего лактат, поскольку пациенты с дисфункцией печени не в состоянии его усвоить. Следует избегать пропофола во время анестезии вследствие его потенциального вмешательства в функцию митохондрий.
У пациентов с лактоацидозом во время разл. процедур следует контролировать уровень лактата. Важно поддерживать анаболизм, для этого рекомендуется сбалансированное потребление жиров и углеводов, ограничение высокоскоростного в/в-введения декстрозы («Глюкозы»), исключение голодания периодом >12 ч.