а) Врожденные ошибки развития. Гены, вызывающие синдромы пороков развития (а также гены, экспрессия которых нарушается под действием факторов окружающей среды или тератогенов), м.б. частью многочисленных клеточных процессов, включая эволюционно консервативные пути передачи сигналов, факторы транскрипции или регуляторные белки, необходимые для ключевых этапов развития. Когда мальформации рассматриваются как изменения, возникающие в результате нарушений важных путей развития, это обеспечивает молекулярную основу для понимания ВПР.
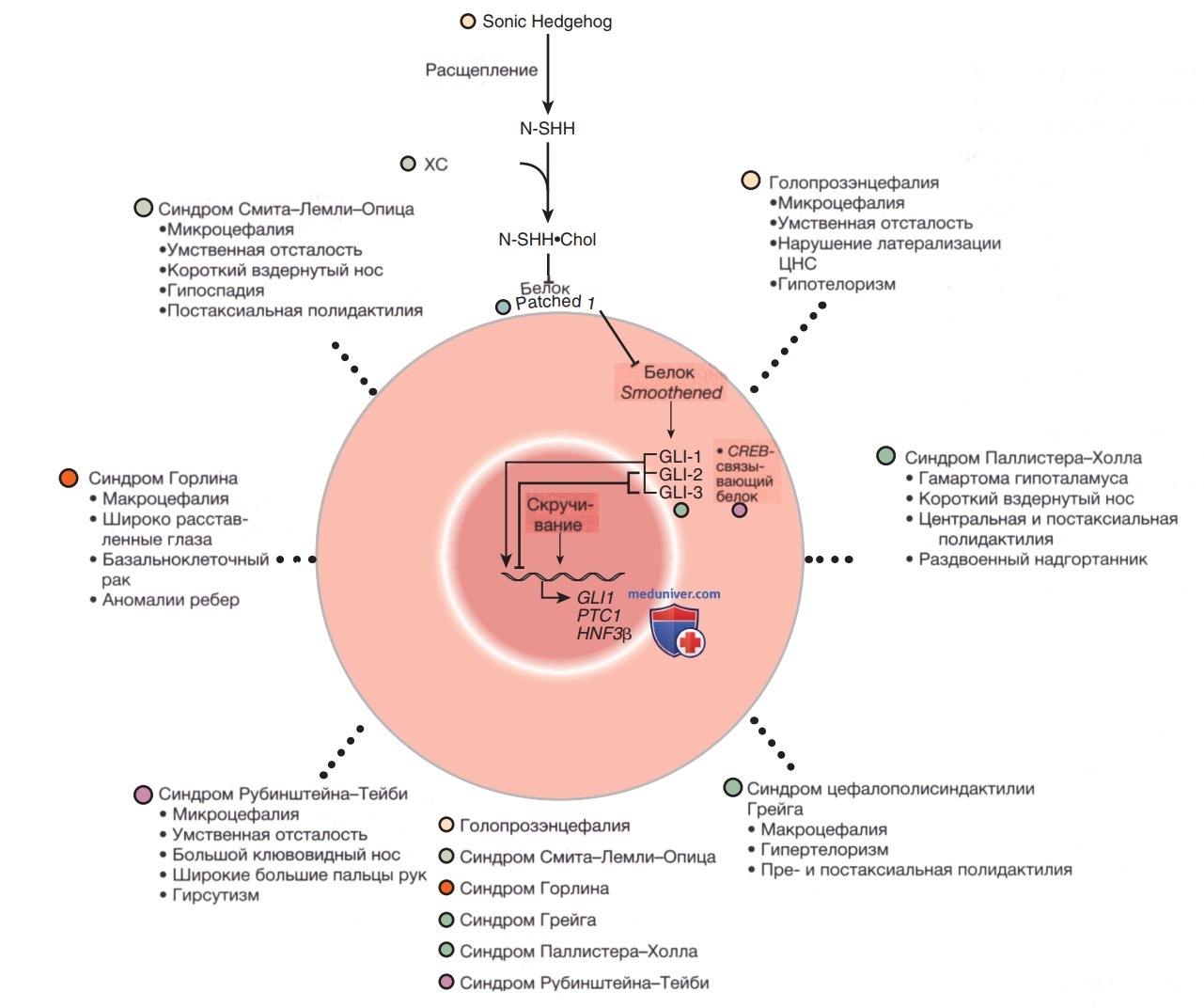
б) Путь белка Sonic Hedgehog в качестве модели. Путь SHH важен для развития во время эмбриогенеза, так как он индуцирует контролируемую пролиферацию тканеспецифическим образом; нарушение определенных этапов этого пути приводит к множеству связанных патологий и ВПР (см. рис. 2). Активация этого пути у взрослых приводит к аномальной пролиферации ткани и раку. Путь SHH преобразует внешний сигнал в форме лиганда в изменения транскрипции гена за счет связывания лиганда со специфическими клеточными рецепторами. SHH — это лиганд, экспрессирующийся у эмбриона в областях, важных для развития ГМ, лица, конечностей и кишечника.
Рисунок 2. Патогенные варианты последовательностей генов, которые функционируют совместно в ходе развития, обычно имеют сходные клинические проявления. Были идентифицированы несколько компонентов пути Sonic Hedgehog (Sonic Hedgehog — семейство генов и соответствующих им белков, управляющих эмбриональным развитием НС и скелетной системы организма, а также выполняющих ряд др. физиол. функций. Белки, кодируемые этими генами, оказывают влияние на сегментацию тела дрозофилы. При выключении гена по всему телу мухи развиваются мелкие шипики. Гиперактивация гена вызывает развитие медуллобластмы ГМ у детей и рака слизистой оболочки рта.) и определены их взаимосвязи (см. более подробную информацию в тексте). Мутации в нескольких членах этого пути приводят к фенотипам с дисморфизмом лица, как при голопрозэнцефалии, синдроме Смита-Лемли-Опица, синдроме Горлина, синдроме цефалополисиндактилии Грейга, синдроме Паллистера-Холла и синдроме Рубинштейна-Тейби
Патогенные варианты последовательности в гене SHH могут вызывать голопрозэнцефалию (см. рис. 2) — дефект средней линии разл. степени тяжести, связанный с разнообразными клиническими эффектами, от циклопии до единственного резца верхней челюсти с гипотелоризмом или близким расстоянием между глазницами. Белок SHH процессируется за счет протеолитического расщепления до активной N-концевой формы, которая затем модифицируется путем добавления холестерина.
Нарушения биосинтеза ХС, в частности гена стерол, 6-7-дегидрохолестеринредуктазы, приводят к синдрому Смита-Лемли-Опица, который также характеризуется голопрозэнцефалией. Модифицированная и активная форма SHH связывается со своим трансмембранным рецептором Patched (PTCH1*).
P.S. * Белок-рецептор РТСН1, который является компонентом сигнального пути Hedgehog, играющего ведущую роль в процессах эмбрионального развития и неопластической трансформации. Мутации гена РТСН1 связаны с развитием синдрома Горлина-Гольца и голопрозэнцефалии.
Связывание SHH с РТСН1 ингибирует активность трансмембранного белка Smoothened (SMO). SMO подавляет активность последующих мишеней пути SHH, семейства транскрипционных факторов GLI, поэтому ингибирование SMO с помощью РТСН1 приводит к активации GLI1, GLI2 и GLI3, что вызывает изменение транскрипции мишеней GLI. РТСН1 и его ортолог, РТСН2, могут действовать как опухолевые супрессоры, а соматические инактивирующие варианты последовательности м.б. связаны с утратой функции опухолевого супрессора, тогда как активирующие мутации в SMO также нередко оказываются онкогенными, особенно при базальноклеточном раке и медуллобластомах.
Инактивирующие мутации зародышевой линии в гене РТСН1 приводят к синдрому Горлина (см. рис. 4) — АуД заболеванию, характеризующемуся дисморфическими чертами (широкое лицо, аномалии зубов, дефекты ребер, короткие пястные кости), базальноклеточными невусами, которые могут подвергаться злокачественной трансформации, и повышенным риском ЗНО, включая медуллобластомы и рабдомиосаркомы. Амплификация гена GLI1 была обнаружена в нескольких опухолях человека, включая глиобластому, остеосаркому, рабдомиосаркому и В-клеточные лимфомы; мутации или изменения в гене GLI3 идентифицированы при синдроме цефалополисиндактилии Грейга, синдроме Паллистера-Холла, постаксиальной полидактилии типа А (и A/В) и преаксиальной полидактилии IV типа (см. рис. 2).
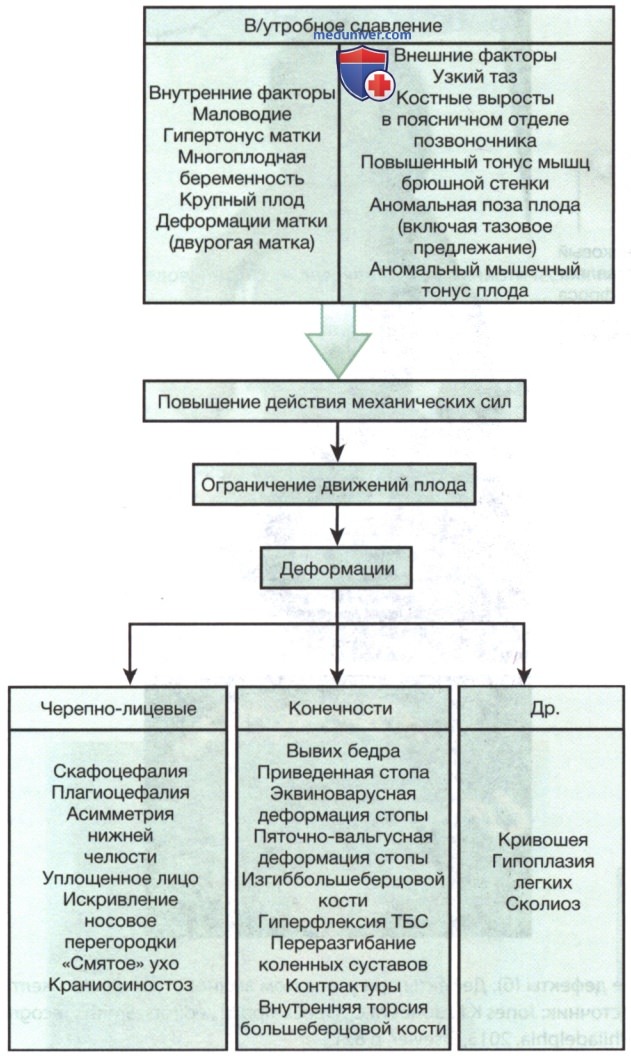
Рисунок 4. Деформационные аномалии, возникающие в результате в/утробного сдавления.
Синдром цефалополисиндактилии Грейга включает гипертелоризм (широко расставленные глаза), синдактилии, преаксиальные полидактилии, а также широкие большие пальцы рук и ног. Синдром Паллистера-Холла — это АуД-заболевание, характеризующееся постаксиальной полидактилией, синдактилией, гамартомами гипоталамуса, атрезией заднего прохода и иногда голопрозэнцефалией. GLI3 связывается с CREB-связывающим белком; при синдроме Рубинштейна-Тейби отмечается гаплонедостаточность этого белка.
Заболевания, вызванные мутациями генов, которые функционируют совместно в пути развития, обычно имеют частично совпадающие клинические проявления. В этом случае общие черты являются результатом экспрессии доменов SHH, которые важны для развития ГМ, лица, конечностей и кишечника. Дефекты ГМ обнаруживаются при голопрозэнцефалии (рис. 9), синдром Смита-Лемли-Опица и синдром Паллистера-Холла. Аномалии лица характерны для голопрозэнцефалии, синдрома Смита-Лемли-Опица, синдрома Горлина, синдрома цефалополисиндактилии Грейга и синдрома Паллистера-Холла.
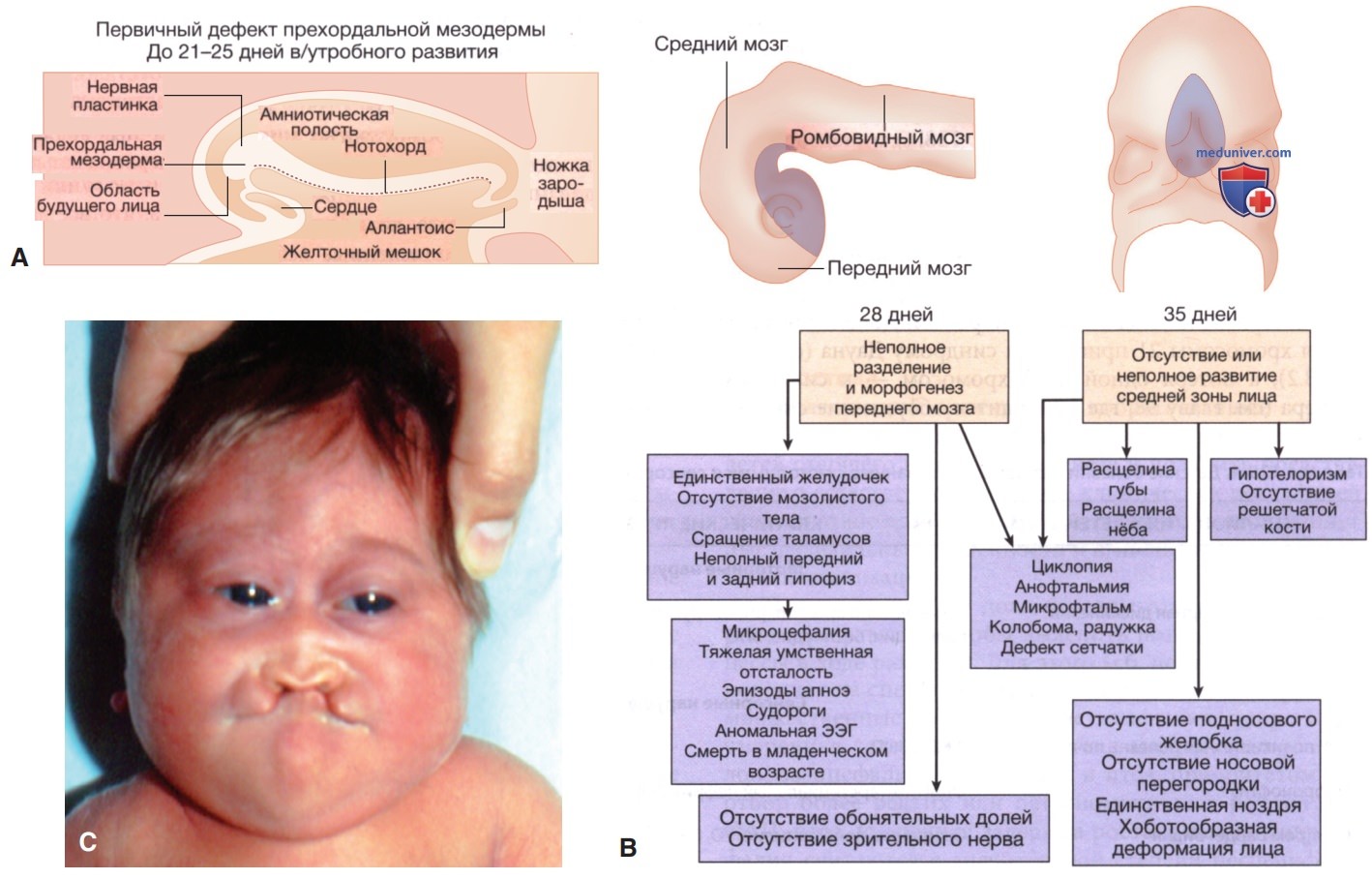
Рисунок 9. Последовательность голопрозэнцефалии: А — схематический продольный разрез 21-дневного эмбриона; В — патогенез развития последовательности; С — ребенок с голопрозэнцефалией.
Дефекты конечностей обнаруживаются при синдроме Смита-Лемли-Опица, синдроме Горлина, синдроме цефалополисиндактилии Грейга, синдроме Паллистера-Холла и синдромах полидактилии. Избыточная экспрессия или активирующие мутации, влияющие на путь SHH, приводят к развитию ЗНО, включая базальноклеточный рак, медуллобластомы, глиобластомы и рабдомиосаркомы.
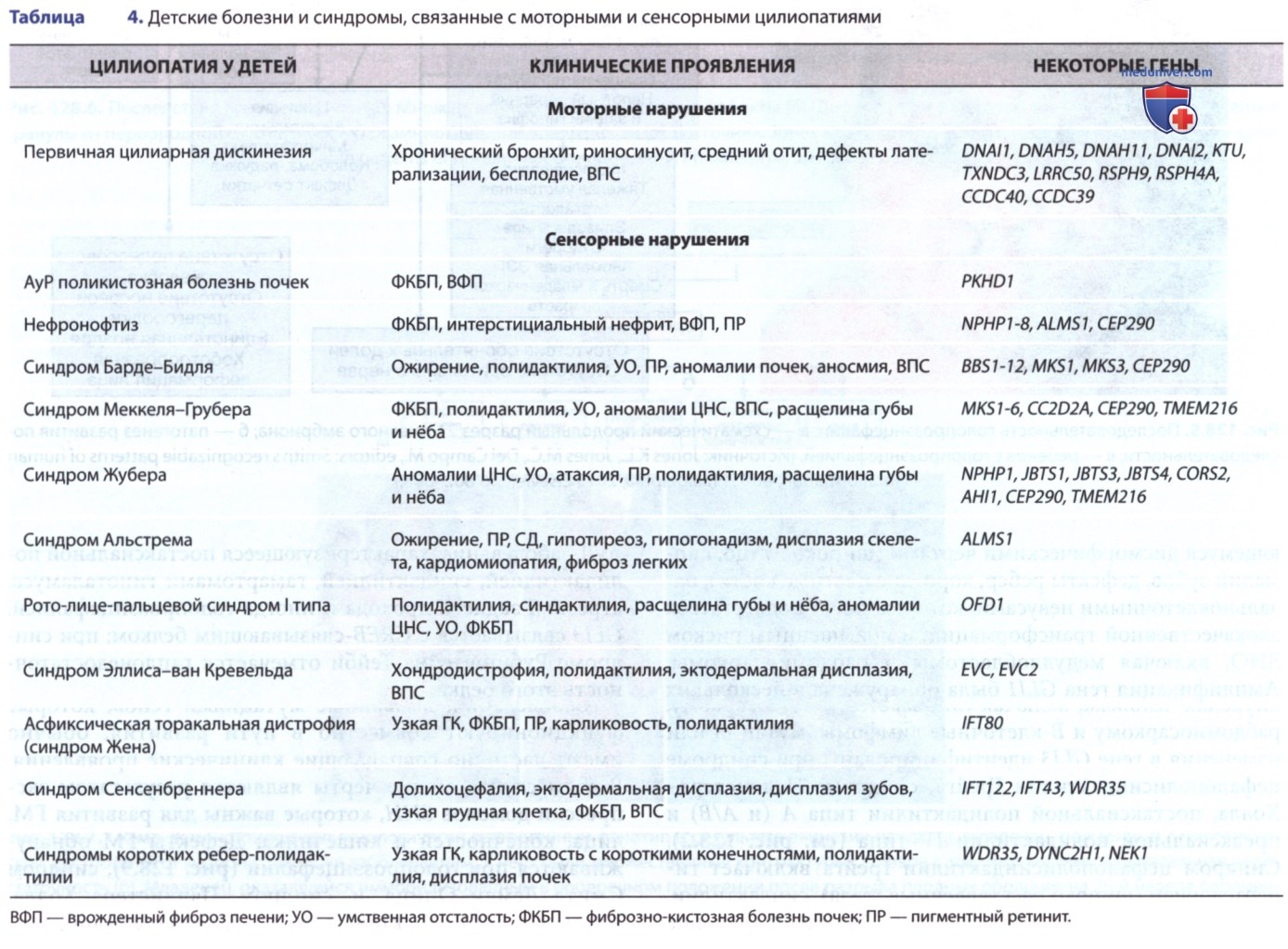
Взаимодействие пути SHH с первичной ресничкой является критическим моментом для передачи внеклеточного сигнала SHH к ядерному аппарату. Ряд заболеваний, включая синдром Барде-Бидля, рото-лице-пальцевой синдром I типа и синдром Жубера, вызваны мутациями в генах, которые обеспечивают функционирование первичной реснички. Эти расстройства, называемые цилиопатиями, клинически совпадают с некоторыми фенотипическими особенностями, описанными ранее, снова демонстрируя, что нарушения консервативных путей развития могут вызывать сходные проявления (табл. 4).
в) Цитогенетические аберрации и хромосомный дисбаланс. Цитогенетический дисбаланс, возникающий из-за дополнительной копии всей хромосомы человека, может привести к характерным и узнаваемым синдромам. Дополнительная копия хромосомы 21 приводит к синдрому Дауна; а потеря одной из Х-хромосом — к синдрому Тернера (см. отдельную статью на сайте (просим Вас пользоваться формой поиска по сайту выше), где приводится обсуждение синдромов с хромосомным дисбалансом в целом).
С появлением цитогенетических методов высокого разрешения, таких как флуоресцентная гибридизация, матричная сравнительная геномная гибридизация и матричный анализ однонуклеотидных полиморфизмов, стало возможным идентифицировать субмикроскопические делеции и дупликации хромосом. Выявлен ряд повторяющихся делеций и дупликаций, вызывающих характерные и узнаваемые синдромы, включая синдром Уильямса (делеция хромосомы 7q11.23), синдром Миллера-Дикера (делеция хромосомы 17р13.3), синдром Смит-Магенис (делеция хромосомы 17р11.2) и синдром делеции 22q11 (делеция хромосомы 22q11.2, также известная как велокардиофациальный синдром/синдром ДиДжорджи).
Матричная сравнительная геномная гибридизация и матричный анализ однонуклеотидных полиморфизмов также позволили выявить более редкие микроделеции и микродупликации, связанные с ВПР, умственной отсталостью и психоневрологическими расстройствами.
Чувствительность и специфичность хромосомных микроматричных анализов сделали этот метод предпочтительным для первоначального обследования ребенка с множественными ВПР и/или умственной отсталостью, хотя важно отметить, что все люди могут иметь многочисленные небольшие микроделеции и микродупликации как нормальные или семейные вариации. Поэтому важно сравнить вариации числа копий у этих детей с ВПР с результатами хромосомного анализа их родителей и с базами данных нормальных вариантов, обнаруженных у лиц без таких врожденных аномалий.