P.S. Клинические рекомендации (КР) в РФ по данной проблеме здравоохранения отсутствуют.
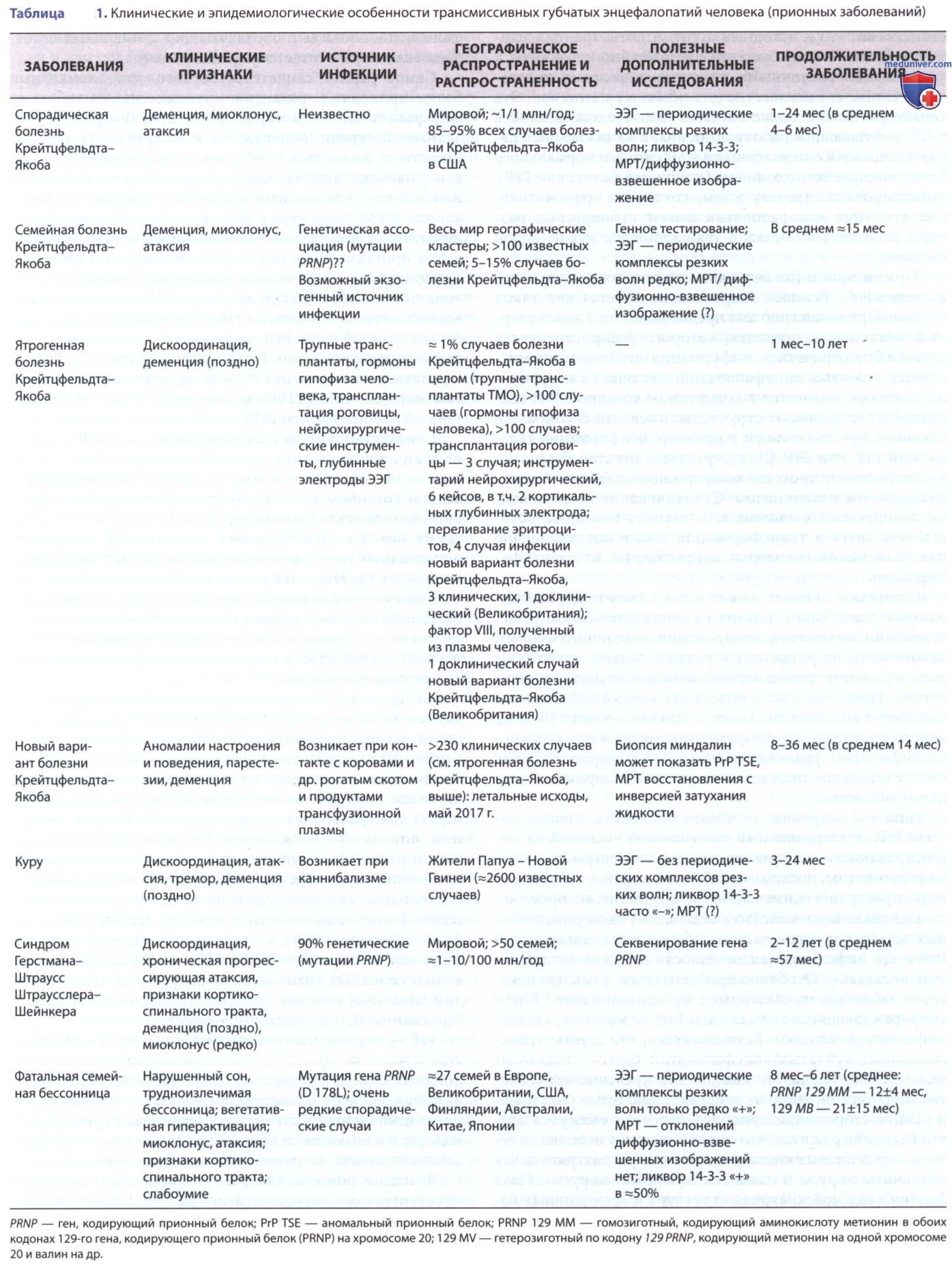
Трансмиссивные губчатые энцефалопатии (TSE; англ. Transmissible spongiform encephalopathies, прионные заболевания) — медленные инфекции НС человека. Существует 4 заболевания (табл. 1): куру; болезнь Крейтцфельдта-Якоба (CJD; англ. Creutzfeldt-Jakob disease) с ее вариантами — спорадической CJD (sCJD), семейной CJD (fCJD), ятрогенной CJD (ятрогенная болезнь Крейтцфельдта-Якоба) и новым вариантом или вариантом CJD (новый вариант болезни Крейтцфельдта-Якоба); синдром Герстманна-Штраусслера-Шейнкера (GSS; англ. Gerstmann-Straussler-Scheinker syndrome) и фатальная семейная бессонница (FFI; англ. Fatal familial insomnia), или синдром спорадической фатальной бессонницы.
Трансмиссивные губчатые энцефалопатии также выявляют у животных. Наиболее распространенные и известные заболевания у животных — скрепи у овец, губчатая энцефалопатия крупного рогатого скота (коровье бешенство) у крупного рогатого скота и хроническая болезнь истощения оленей и лосей, встречающаяся в некоторых частях США, Канады, Норвегии и Финляндии. Все трансмиссивные губчатые энцефалопатии имеют сходные клинические и гистопатологические проявления, и все они считаются медленными инфекциями с очень длительными бессимптомными инкубационными периодами (часто годами), продолжительностью несколько месяцев или более. Заболевание поражает только НС.
Трансмиссивные губчатые энцефалопатии неуклонно прогрессируют после начала болезни и неизменно приводят к летальному исходу. Наиболее ярким невропатологическим изменением, которое происходит при каждой трансмиссивной губчатой энцефалопатии, считается губчатая дегенерация серого вещества коры ГМ.
а) Этиология. Трансмиссивные губчатые энцефалопатии передаются восприимчивым животным при инокуляции тканей пораженных субъектов. Хотя инфекционные агенты реплицируются в некоторых культурах клеток, они не достигают высоких титров инфекционности, обнаруживаемых в тканях ГМ, и не вызывают заметных цитопатических эффектов в культурах. В большинстве предыдущих исследований агентов трансмиссивных губчатых энцефалопатий использовались тесты in vivo, основанные на передаче типичного неврологического заболевания животным в качестве доказательства того, что агент присутствовал и был неповрежденным.
Инокуляция восприимчивых животных-реципиентов небольшими количествами инфекционного агента трансмиссивных губчатых энцефалопатий через несколько месяцев приводит к накоплению в тканях большого количества агента с такими же физическими и биологическими свойствами, что и исходный агент. Агенты трансмиссивных губчатых энцефалопатий чрезвычайно устойчивы к инактивации различными хим. и физическими методами лечения, что неизвестно для обычных патогенов. Эта характеристика, а также их частичная чувствительность к ЛП, нарушающим работу протеина, и их последовательная ассоциация с аномальными изоформами нормального белка, кодируемого хозяином (прионный белок, или PrP), стимулировали гипотезу о том, что агенты трансмиссивных губчатых энцефалопатий имеют субвирусные размеры и состоят из белка, а также лишены нуклеиновой кислоты.
Термин прион (от белкового инфекционного агента), введенный С. Prusiner, широко используется для таких агентов. Прионная гипотеза предполагает, что молекулярный механизм, с помощью которого распространяется патоген-специфическая информация агентов трансмиссивных губчатых энцефалопатий, включает самореплицирующееся изменение в кодируемом хозяином PrP, связанное с переходом от структуры, имеющей α-спираль в нативной чувствительной к протеазе конформации (клеточный PrP или PrP С), к структуре, богатой β-слоями, в устойчивой к протеазе конформации, связанной с инфекционным потенциалом. Существование второго белка, кодируемого хозяином, называемого белком X, который участвует в трансформации, также постулировано для объяснения некоторых открытий, но не идентифицировано.
Гипотеза о прионах до сих пор не получила всеобщего признания. Она основана на постулированном существовании механизма кодирования, подобного геному, основанного на различиях в укладке белков, которые не нашли удовлетворительного объяснения на молекулярном уровне. Предстоит также объяснить многие наблюдаемые биологические штаммы агента трансмиссивных губчатых энцефалопатий, хотя обнаружены и предложены штаммоспецифические различия в аномальных формах PrP как обеспечивающие правдоподобную молекулярную основу для кодирования.
Гипотеза о прионах не может объяснить, почему чистый PrP, не загрязненный нуклеиновой кислотой от инфицированного хозяина, не передал типичную губчатую энцефалопатию, последовательно связанную с серийно самораспространяющимся агентом. Открытие, которое также вызывало беспокойство в нескольких экспериментальных моделях и человеческих заболеваниях: аномальный PrP и его инфекционная активность не были последовательно связаны. Особенно проблематичен факт, что некоторые заболевания, связанные с мутациями в гене PRNP и сопровождающиеся аномальным PrP, не могут передавать инфекцию животным.
Если окажется, что агенты трансмиссивных губчатых энцефалопатий состоят только из белка без обязательного компонента нуклеиновой кислоты, тогда термин «прион» действительно будет уместным, и ранние сторонники прионной гипотезы окажутся правы. Если обнаружится, что агенты содержат небольшие геномы нуклеиновых кислот, то их лучше рассматривать как атипичные вирусы и называть термином «вирусы».
Пока фактическая молекулярная структура инфекционных патогенов трансмиссивных губчатых энцефалопатий и наличие или отсутствие генома нуклеиновой кислоты не будут строго установлены, менее спорно называть их агентами трансмиссивных губчатых энцефалопатий. Большинство авторитетных источников приняло термин «прион» (иногда относящийся к агенту трансмиссивных губчатых энцефалопатий, а иногда и обозначающий аномальный белок, даже если он не считается передаваемым).
Самое раннее свидетельство того, что аномальные белки приводят к развитию трансмиссивных губчатых энцефалопатий, было морфологическим. Фибриллы, связанные со скрепи, обнаружены в экстрактах тканей пациентов и животных с губчатой энцефалопатией, но не в нормальных тканях. Связанные со скрепи фибриллы напоминают амилоидные фибриллы, которые накапливаются в ГМ пациентов с болезнью Альцгеймера, но отличаются от них. Группа АГн-связанных белков, устойчивых к протеазе (PrP), оказалась компонентами фибрилл, ассоциированных со скрепи. Она присутствовала в амилоидных бляшках, обнаруженных в ГМ пациентов и животных с трансмиссивными губчатыми энцефалопатиями.
Аномальные формы PrP по-разному обозначаются разными авторитетами как PrP Sc (PrP типа скрепи), PrP-res (протеазорезистентный PrP), PrP трансмиссивных губчатых энцефалопатий (TSE-связанный PrP) или PrP D (связанный с заболеванием PrP).
Неизвестно, составляет ли аномальный PrP полную инфекционную частицу губчатой энцефалопатии, считается ли он компонентом этих частиц или патологическим белком-хозяином, не отделенным от фактического инфекционного объекта с помощью используемых в настоящее время методов. Демонстрация того, что PrP кодируется нормальным геном-хозяином, подтверждает последнюю возможность. Несколько исследований показывают, что специфическая для агента патогенная информация может передаваться и воспроизводиться различными конформациями белка с одной и той же первичной аминокислотной последовательностью в отсутствие специфичных для агента нуклеиновых кислот.
Обнаружено, что свойства двух грибных белков передаются по наследству без кодирования нуклеиновой кислотой, но не передаются естественным образом грибам-реципиентам в качестве инфекционных элементов. Каким бы ни было его отношение к реальным инфекционным частицам трансмиссивных губчатых энцефалопатий, PrP играет центральную роль в восприимчивости к инфекции, потому что нормальный PrP должен экспрессироваться у мышей и крупного рогатого скота для поддержания репликации инфекционных агентов. Унаследованные нормальные вариации фенотипа PrP приводят к повышенной восприимчивости к новому варианту болезни Крейтцфельдта-Якоба и (в меньшей степени) к спорадической болезни Крейтцфельдта-Якоба и к возникновению семейных трансмиссивных губчатых энцефалопатий (семейная болезнь Крейтцфельда-Якоба и синдрома Герстманна-Штраусслера-Шейнкера).
PrP — гликопротеины; протеазорезистентные PrP при агрегировании обладают физическими свойствами амилоидных белков. PrP разных видов животных похожи по аминокислотным последовательностям и антигенности, но не идентичны по структуре. Первичная структура PrP кодируется хозяином и не изменяется источником инфекционного агента, вызывающего его образование.
Функция повсеместно распространенного протеазочувствительного предшественника PrP (обозначенного PrP С для клеточного PrP или PrP-sen для протеазочувствительного PrP) в нормальных клетках неизвестна. Он связывает медь и может играть определенную роль в нормальной синаптической передаче. Он не требуется для жизни или для относительно нормальной мозговой функции у мышей и крупного рогатого скота. Животные должны экспрессировать PrP для развития болезни скрепи и для поддержки репликации агентов трансмиссивных губчатых энцефалопатий.
Степень гомологии между аминокислотными последовательностями PrP у разных видов животных может коррелировать с видовым барьером, который влияет на восприимчивость животных одного вида к инфекции агентом трансмиссивных губчатых энцефалопатий, адаптированным для роста в др. виде. Степень гомологии последовательностей не всегда может прогнозировать восприимчивость к одному и тому же агенту трансмиссивных губчатых энцефалопатий.
Попытки найти частицы, похожие на частицы вирусов или вирусоподобных агентов, в тканях ГМ людей или животных с губчатой энцефалопатией не увенчались успехом. Своеобразные тубуловезикулярные структуры, напоминающие некоторые вирусы, наблюдались в тонких срезах тканей ГМ, инфицированных агентами трансмиссивных губчатых энцефалопатий, и культивируемых клеток, но не в нормальных клетках. Не было установлено, что эти структуры влияют на инфекционный потенциал агентов.
б) Эпидемиология. Куру поразил многих детей обоего пола >4 лет, подростков и молодых людей (в основном женщин), живущих в одном ограниченном районе Папуа — Новой Гвинеи. Полное исчезновение куру у людей, родившихся после 1957 г., позволяет сделать выод, что практика ритуального каннибализма (которая закончилась в том же году) была единственным механизмом распространения инфекции в Папуа - Новой Гвинее.
Раньше считалось, что болезнь Крейтцфельдта-Якоба, наиболее распространенная губчатая энцефалопатия человека, встречается только у пожилых людей; но ятрогенная и спорадическая болезнь Крейтцфельдта-Якоба поражала и молодых людей (7 случаев у подростков, один — у девочки 14 лет). Единственный случай спорадической бессонницы со смертельным исходом выявлен у американского подростка. Болезнь Крейтцфельдта-Якоба не диагностируют у детей и подростков, но новый вариант болезни Крейтцфельдта-Якоба чаще возникает у молодых людей. Из 174 случаев нового варианта болезни Крейтцфельдта-Якоба, зарегистрированных за 2010 г. в Соединенном Королевстве, все, за исключением 23 случаев, отмечены у людей <40 лет и 22 — у людей <20 лет.
Самый молодой возраст начала заболевания — 12 лет. Спорадическая болезнь Крейтцфельдта-Якоба признана во всем мире с ежегодными показателями 0,25-2:1000 000 населения (без учета возраста).
При этом очаги болезни Крейтцфельдта-Якоба значительно чаще встречаются среди ливийских евреев в Израиле, в изолированных деревнях Словакии и в др. ограниченных областях. Спорадическая болезнь Крейтцфельдта-Якоба не вызвана каким-либо обычным контактом с зараженным объектом, и источник инфекции остается неизвестным. Сторонники прионной гипотезы убеждены, что белок РгР может спонтанно неправильно складываться, получает способность к саморепликации и вызывает спорадическую болезнь Крейтцфельдта-Якоба. Скептики отдают предпочтение заражению неизвестным повсеместно распространенным агентом трансмиссивных губчатых энцефалопатий с низкой скоростью атаки, за исключением людей с определенными мутациями в гене PRNP. Ни одна из этих возможных этиологий не доказана. Передача от человека к человеку подтверждена только для ятрогенных случаев.
Супруги и домашние контакты пациентов не подвержены риску заражения болезнью Крейтцфельдта-Якоба, хотя сообщалось о двух случаях заболевания среди супругов. Мед. персонал, контактирующий с мозгом пациентов с болезнью Крейтцфельдта-Якоба, подвержен повышенному риску заражения. У ~20 мед. работников диагностировано это заболевание.
Сходство болезни Крейтцфельдта-Якоба со скрепи вызвало опасения, что инфицированные ткани овцы м.б. источником губчатой энцефалопатии у людей. Нет надежных эпидемиологических данных, свидетельствующих о том, что воздействие потенциально зараженных скрепи животных, мяса, мясных продуктов или экспериментальных препаратов возбудителя приводит к передаче трансмиссивных губчатых инфекций человеку. Инфекционный потенциал агента хронической болезни истощения в отношении людей также не продемонстрирован, но все еще исследуется. Олени, лоси и лоси в 15 штатах США и 2 провинциях Канады инфицированы естественным путем; случаи хронической болезни истощения обнаружены у диких северных оленей и лосей (европейских лосей) в Норвегии и Финляндии.
Потребление зараженного мяса, включая оленину от животных, инфицированных возбудителем хронической болезни истощения, не считалось фактором риска трансмиссивных губчатых инфекций человека в эпидемиологических исследованиях. Недавнее неопубликованное исследование, для которого потребовалось несколько лет, предоставило доказательства, что хроническая болезнь истощения экспериментально передавалась обезьянам, которых кормили олениной от клинически здоровых инфицированных оленей. Это побудило канадские власти дать рекомендации по охране здоровья. Вспышка губчатой энцефалопатии среди крупного рогатого скота (инфицированного в результате употребления в пищу мясокостной муки, зараженной скрепи и добавленной в корм) впервые обнаружена в Соединенном Королевстве в 1986 г., а позже зарегистрирована у крупного рогатого скота в 27 др. странах, включая Канаду и США.
Всемирная организация здравоохранения животных (OIE) сообщила о 190 000 случаев губчатой энцефалопатии крупного рогатого скота, почти 97% из них — из Соединенного Королевства. Число случаев заболевания в Соединенном Королевстве постепенно уменьшалось после 1992 г., а затем и в др. странах. В 2016 г. зарегистрировано только 2 случая во всем мире (из Франции и Испании) и ни одного случая из Соединенного Королевства. Обнаружение нового агента трансмиссивных губчатых энцефалопатий у животных семейства копытных и кошачьих в британских зоопарках, а затем и у домашних кошек, вызвало опасения, что агент губчатой энцефалопатии крупного рогатого скота приобрел более широкий диапазон восприимчивых хозяев, что представляет потенциальную опасность для человека.
Это остается наиболее правдоподобным объяснением эпизодов губчатой энцефалопатии крупного рогатого скота, впервые описанных у подростков в Великобритании в 1996 г. и, по состоянию на май 2017 г., в конечном итоге затронувшего 178 человек в Соединенном Королевстве (не считая тревожного числа людей с признаками возможной бессимптомной или «доклинической» инфекции новый вариант болезни Крейтцфельдта-Якоба) и >50 человек в 11 др. странах (всего 231 случай во всем мире): 27 во Франции, 5 в Испании, 4 в Ирландии, 3 в Нидерланды, по 2 случая в Италии и Португалии, а также единичные случаи в Японии и Саудовской Аравии. Новый вариант болезни Крейтцфельдта-Якоба также встречался у бывших жителей Великобритании (>6 мес), проживающих в Ирландии (2 случая), Франции (1 случай), Канаде (1 случай), Тайване (1 случай) и США (2 случая).
Два случая нового вариант болезни Крейтцфельдта-Якоба — 1 в США и 1 в Канаде — зарегистрированы у бывших постоянных жителей Саудовской Аравии, страны, которая не признала заболевание, но могла импортировать зараженные мясные продукты из Соединенного Королевства. Третий случай нового варианта болезни Крейтцфельдта-Якоба ранее был подтвержден у гражданина Саудовской Аравии, проживающего в Саудовской Аравии. Самый последний случай нового варианта болезни Крейтцфельдта-Якоба, диагностированный в США, произошел у иммигранта, который, по мнению CDC, был инфицирован в первые годы, проведенные в Кувейте.
Ни один случай нового варианта болезни Крейтцфельдта-Якоба не был подтвержден у человека, родившегося в Соединенном Королевстве после 1989 г. Однако исследование резецированных аппендиксов в Соединенном Королевстве на предмет наличия субклинической инфекции прионами показало, что у 1:2000 обследованных людей обнаружено накопление PrP в лимфоидных фолликулах. Остается спорным, были ли эти накопления результатом субклинического течения нового варианта болезни Крейтцфельдта-Якоба или др. трансмиссивной губчатой энцефалпатии. Ни один из субъектов на сегодняшний день не обращался за МП с явными проявлениями трансмиссивной губчатой энцефалопатии.
Ятрогенная передача болезни Крейтцфельдта-Якоба выявлена >30 лет назад (табл. 2). Случайные передачи болезни Крейтцфельдта-Якоба возможны при использовании зараженных нейрохирургических инструментов (с 1980 г. не зарегистрировано ни одного случая) или операционных помещений, кортикальных электродов, загрязненных во время операции по поводу эпилепсии, инъекций человеческого гормона роста трупного гипофиза и гонадотропина (больше не продаются в США), а также при трансплантации зараженных роговиц и аллотрансплантатов ТМО человека, которые все еще ограниченно используются в США в качестве хирургического пластыря. Фармацевтические ЛП и тканевые трансплантаты, полученные из нервных тканей человека или загрязненные ими, представляют особый риск (особенно если получены от сомнительных доноров и больших групп доноров).
Исследования животных, экспериментально инфицированных агентами трансмиссивной губчатой энцефалпатии, впервые показали, что кровь и компоненты крови людей с доклинической инфекцией м.б. факторами риска передачи болезни реципиентам. С 1980-х гг. такие компоненты крови изъяты в качестве меры предосторожности в США, когда у донора выявили болезнь Крейтцфельдта-Якоба, а ЛП его крови все еще использовались. В рамках Программы наблюдений в Соединенном Королевстве сообщалось о новом варианте болезни Крейтцфельдта-Якоба у трех реципиентов не лейкоредуцированных эритроцитов от доноров. У доноров позже был диагностирован новый вариант болезни Крейтцфельдта-Якоба.
Было проведено вскрытие у четвертого реципиента эритроцитов, умершего от др. заболевания (новый вариант болезни Крейтцфельдта-Якоба не встречался ни у одного реципиента лейкоцитов, полученных от донора, у которого позже развился новый вариант болезни Крейтцфельдта-Якоба).
Исследование, проводимое в течение 20 лет Американским Красным Крестом и CDC, не обнаружило реципиента компонентов крови, полученных от доноров, у которых позже были диагностированы спорадические заболевания.
Доказательства доклинической инфекции нового варианта болезни Крейтцфельдта-Якоба обнаружены при вскрытии у пациента из Великобритании с гемофилией А, получавшего человеческий фактор свертывания крови VIII из плазмы крови (от одного донора с новым вариантом болезни Крейтцфельдта-Якоба). Задействованный фактор свертывания крови не был лицензирован в США. Власти Великобритании описали два случая спорадической болезни Крейтцфельдта-Якоба у реципиентов факторов свертывания крови, полученных из плазмы (у обоих в анамнезе также были переливания компонентов крови). Ученые отметили, что это открытие м.б. случайным.
в) Патология и патогенез. Возможные входные ворота для агента куру — ЖКТ или повреждения на слизистой оболочки рта или коже, случайно подвергшиеся воздействию агента во время каннибализма. Считается, что пациенты с новым вариантом болезни Крейтцфельдта-Якоба (и животные губчатой энцефалопатией крупного рогатого скота) также были инфицированы агентом губчатой энцефалопатии крупного рогатого скота при употреблении загрязненных продуктов из говядины. За исключением прямого введения в НС, первый участок репликации агентов трансмиссивных губчатых энцефалопатий находится в тканях РЭС.
Агенты трансмиссивных губчатых энцефалопатий обнаружены в низких титрах в крови экспериментально инфицированных животных (мышей, обезьян, хомяков и овец и в крови людей с новым вариантом болезни Крейтцфельдта-Якоба и спорадической болезнью Крейтцфельдта-Якоба). Циркулирующие лимфоидные клетки необходимы для заражения мышей периферическими путями. Ограниченные данные свидетельствуют о том, что агенты трансмиссивных губчатых энцефалопатий также распространяются в ЦНС по восходящим периферическим нервам. Несколько исследовательских групп заявили об обнаружении возбудителя болезни Крейтцфельдта-Якоба в крови человека, хотя др. исследователи потерпели неудачу.
При куру единственный выход агента из организма в количествах, достаточных для заражения др. людей, — через инфицированные ткани во время каннибализма. При ятрогенно передающейся болезни Крейтцфельдта-Якоба ГМ и глазные яблоки пациентов были вероятными источниками заражения. Экспериментальная передача агента животным через почки, печень, легкие, ЛУ и селезенку показала, что эти ткани и СМЖ содержат инфекционный агент болезни Крейтцфельдта-Якоба. Ни один из этих источников не был причастен к случайной передаче болезни Крейтцфельдта-Якоба человеку. Не выявлено АТл или клеточного иммунитета к инфекционным агентам у пациентов и животных. Однако мыши должны быть иммунологически компетентными, чтобы периферическими путями инокуляции заразиться возбудителем скрепи.
Типичные изменения при трансмиссивных губчатых инфекциях — вакуолизация и потеря нейронов с гипертрофией и пролиферацией глиальных клеток. Они наиболее выражены в коре ГМ у пациентов с болезнью Крейтцфельдта-Якоба и в мозжечке у пациентов с куру. Поражения ЦНС затрагивают серое в-во или даже ограничиваются им на ранней стадии заболевания. Потеря миелина вторична по отношению к дегенерации нейронов. Воспаление отсутствует, но часто наблюдается заметное увеличение количества и размера астроцитов. Губчатые изменения не считаются редкой находкой на вскрытии у пациентов с фатальной семейной бессоницей, а дегенерация нейронов и глиоз ограничены ядрами таламуса.
Амилоидные бляшки обнаруживаются в ГМ всех пациентов с синдромом Герстмана-Штраусслера-Шейнкера и у 70% пациентов с куру. Эти бляшки реже встречаются у пациентов с болезнью Крейтцфельдта-Якоба. Амилоидные бляшки чаще встречаются в мозжечке, но образуются и в др. частях мозга. В ГМ пациентов с новым вариантом болезни Крейтцфельдта-Якоба бляшки, окруженные ореолом вакуолей (описываемые как «цветочные» или «цветущие бляшки»), встречаются часто. Амилоидные бляшки при трансмиссивных губчатых энцефалопатиях реагируют с антисывороткой, приготовленной против PrP. Даже при отсутствии бляшек внеклеточный PrP можно обнаружить в паренхиме ГМ с помощью иммуноокрашивания.
г) Клинические проявления. Куру (ныне ликвидировано) — прогрессирующее дегенеративное заболевание мозжечка и ствола ГМ с менее очевидным поражением коры ГМ. Первый признак куру — мозжечковая атаксия с последующим прогрессирующим нарушением координации. Характерны грубые дрожащие толчкообразные движения конечностей, различные нарушения функции ЧМН, часто с нарушением сопряженного взгляда и глотания. Пациенты умирали от истощения и пневмонии или от ожогов (во время готовки пищи) в течение года после начала болезни. Часто отмечались изменения в мышлении, но не было явной деменции или прогрессирования до комы, как при болезни Крейтцфельдта-Якоба. Признаков острого энцефалита в виде лихорадки, головных болей и судорог не было.
Болезнь Крейтцфельдта-Якоба выявляется по всему миру. Пациенты изначально страдают либо сенсорными нарушениями (чаще зрительными), либо спутанностью сознания и несоответствующим поведением, которые в течение недель или месяцев прогрессируют до явной деменции, акинетического мутизма и комы. У некоторых пациентов мозжечковая атаксия наблюдается на ранней стадии заболевания, и у большинства пациентов отмечены миоклонические подергивания. Среднее время выживания пациентов со спорадической болезнью Крейтцфельдта-Якоба — <1 года с момента появления первых признаков заболевания, хотя 10% живут в течение 2 лет.
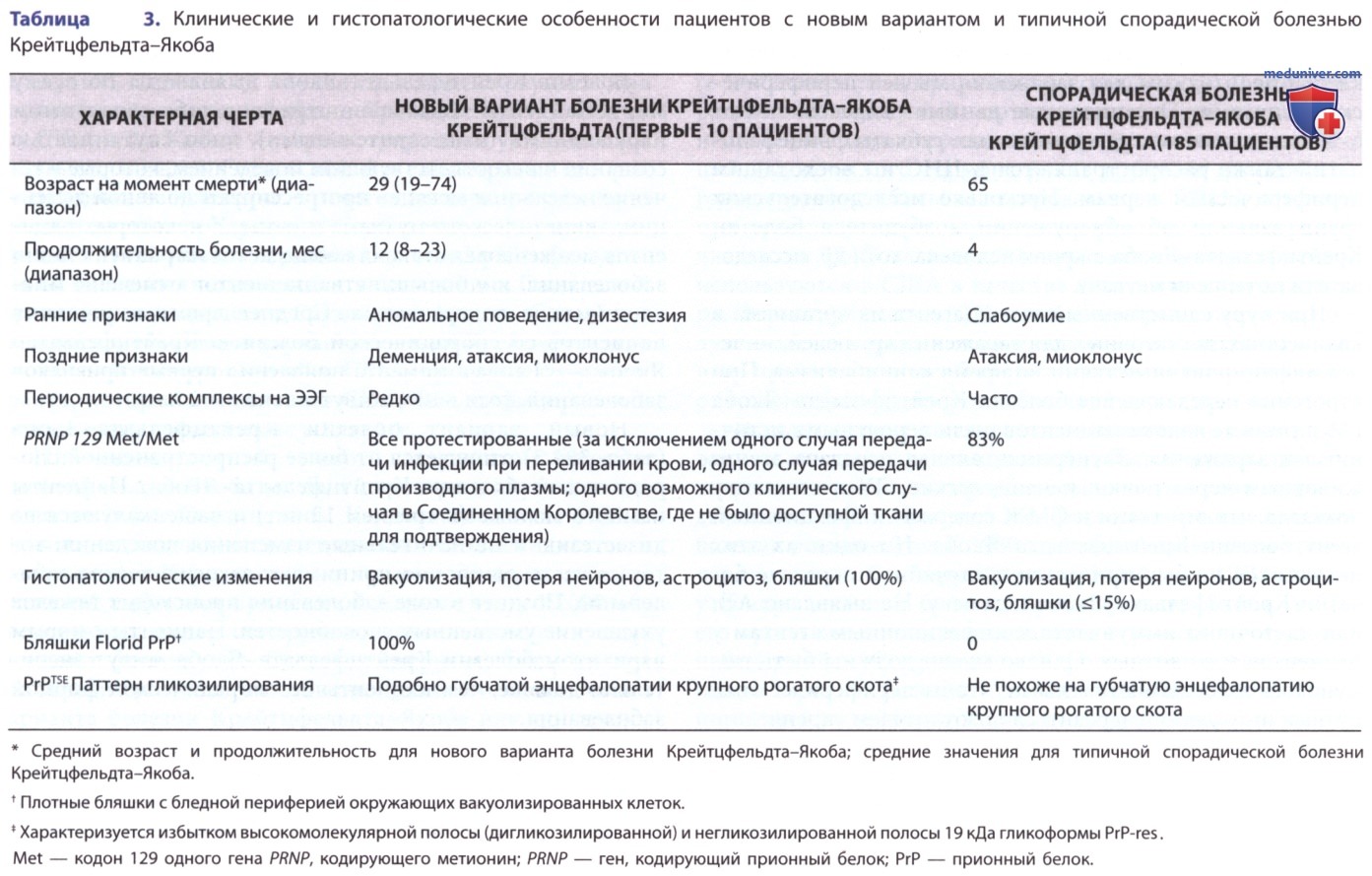
Новый вариант болезни Крейтцфельдта-Якоба (табл. 3) отличается от более распространенной спорадической болезни Крейтцфельдта-Якоба. Пациенты намного моложе (в среднем 12 лет) и чаще жалуются на дизестезию и незначительные изменения поведения, которые часто ошибочно принимают за психическое заболевание. Позднее в ходе заболевания происходит тяжелое ухудшение умственных способностей. Пациенты с новым вариантом болезни Крейтцфельдта-Якоба живут значительно дольше, чем пациенты со спорадической формой заболевания.
Предприняты попытки классифицировать случаи болезни Крейтцфельдта-Якоба на основе электрофоретических различий в PrPTSE и вариаций его чувствительности к перевариванию протеиназой протеолитического фермента (РК). Для разных вариантов типичны различные клинические характеристики, включая продолжительность болезни, хотя все трансмиссивные губчатые энцефалопатии в конечном итоге фатальны.
Синдром Герстмана-Штраусслера-Шейнкера — семейное заболевание, напоминающее болезнь Крейтцфельдта-Якоба, но с более выраженной мозжечковой атаксией и амилоидными бляшками. Деменция может появиться только на поздних стадиях, а средняя продолжительность заболевания больше, чем у спорадической болезни Крейтцфельдта-Якоба. Прогрессирующая тяжелая бессонница и дизавтономия, а также атаксия, миоклонус и др. признаки, напоминающие признаки болезни Крейтцфельдта-Якоба и синдром Герстмана-Штраусслера-Шейнкера, характерны для фатальной семейной бессонницы и спорадической фатальной бессонницы. Описан случай спорадической бессонницы со смертельным исходом у подростка. Синдром Герстмана-Штраусслера-Шейнкера не диагностируется у детей и подростков.
д) Диагностика. Диагноз губчатой энцефалопатии устанавливают на основании клинических данных после исключения др. заболеваний. Присутствие белка 14-3-3 (см. «Лабораторные данные») в СМЖ помогает в ДД болезни Крейтцфельдта-Якоба и болезни Альцгеймера, но не у детей. Повышение уровня белка 14-3-3 в СМЖ не считается специфическим для трасмиссивных губчатых энцефалпатий и часто встречается при вирусном энцефалите и др. состояниях, вызывающих быстрый некроз ткани мозга. Биопсия ГМ м.б. полезна в диагностике болезни Крейтцфельдта-Якоба, но ее можно рекомендовать только в том случае, если предстоит исключить потенциально излечимое заболевание или при наличии др. веских причин для постановки предубойного диагноза у животных.
Для постановки окончательного диагноза требуется микроскопическое исследование ткани ГМ, полученное при вскрытии. Демонстрация протеазоустойчивого PrP в экстрактах мозга подтверждает гистопатологический диагноз. Накопление патологического PrP в лимфоидных тканях даже до появления неврологических симптомов типично для нового варианта болезни Крейтцфельдта-Якоба. Биопсия миндалин поможет избежать биопсии ГМ при подозрении на новый вариант болезни Крейтцфельдта-Якоба.
На сегодняшний день ни один анализ крови не прошел валидацию для прижизненного тестирования людей или животных. Передача болезни восприимчивым животным путем инокуляции суспензии ГМ (чувствительный, специфический и надежный метод) зарезервирована для случаев, представляющих особый исследовательский интерес.
е) Лабораторные данные. Практически у всех пациентов с типичными спорадическими, ятрогенными и семейными формами болезни Крейтцфельдта-Якоба наблюдаются аномальные данные ЭЭГ по мере прогрессирования заболевания: фон становится медленным и неравномерным с уменьшением амплитуды. Могут также появиться различные пароксизмальные явления, такие как медленные волны, резкие волны и спайк-волновые комплексы, и они м.б. односторонними, фокальными или двусторонне синхронными. Пароксизмальные явления м.б. вызваны громким шумом. У многих пациентов наблюдаются типичные периодические комплексы подавления-взрыва высоковольтной медленной активности на ЭЭГ на каком-то этапе болезни.
У пациентов с новым вариантом болезни Крейтцфельдта-Якоба наблюдается только генерализованное замедление, без периодических всплесков высоковольтных разрядов на ЭЭГ. КТ или МРТ могут показать атрофию коры ГМ и большие желудочки на поздних стадиях болезни Крейтцфельдта-Якоба.
У многих пациентов с новым вариантом болезни Крейтцфельдта-Якоба отмечено увеличение плотности ЛА на МРТ. Надежную интерпретацию изображений лучше доверить опытным рентгенологам.
У пациентов с болезнью Крейтцфельдта-Якоба возможно умеренное повышение уровня белка в СМЖ. Необычные белковые пятна наблюдались в образцах СМЖ после двумерного разделения в гелях и окрашивания серебром; пятна были идентифицированы как белки 14-3-3, нормальные белки (не связанные с РгР), которые в изобилии присутствуют в нейронах, но не обнаруживаются в СМЖ. Однако белок 14-3-3 также был обнаружен в образцах СМЖ некоторых пациентов с острыми вирусными энцефалитами и недавними церебральными инфарктами, и он не считается специфичным для болезни Крейтцфельдта-Якоба.
Обнаружение белка 14-3-3 в СМЖ не считается чувствительным и специфическим методом, но полезно в подтверждении диагноза нового варианта болезни Крейтцфельдта-Якоба (особенно при увеличении уровня др. клеточных белков). Диагноз основывается на распознавании типичного сочетания анамнестических данных, клинического течения и обследований (исследование СМЖ, КТ или МРТ, ЭЭГ), подтвержденных гистопатологией и обнаружением PrPTSE в тканях мозга при вскрытии (или, реже, в тканях миндалин или при биопсии ГМ).
ж) Лечение. Ни один вариант терапии не доказал свою эффективность. Исследования культур клеток и грызунов, экспериментально инфицированных агентами трансмиссивных губчатых инфекций, показали, что лечение хлорпромазином, хинакрином и тетрациклинами м.б. полезным, особенно во время инкубационного периода. Результаты клинических испытаний, основанные на этих исследованиях, недостоверны, и кажется маловероятным, что серьезное повреждение ГМ на поздней стадии заболевания можно обратить вспять с помощью ЛП. Инфузии полисульфата пентозана непосредственно в желудочки ГМ замедлили прогрессирование заболевания у одного пациента, но не повлияли на полученное ранее повреждение ГМ.
Всем пациентам с болезнью Крейтцфельдта-Якоба, как и при др. прогрессирующих фатальных неврологических заболеваниях, показана соответствующая поддерживающая терапия. На основе экспериментальных исследований на животных предложено несколько режимов профилактического лечения после контакта, но ни один из них не получил широкого признания.
з) Генетическая консультация. Трансмиссивные губчатые энцефалопатии иногда возникают в семьях по АуД-типу наследования. У пациентов с семейным анамнезом болезни Крейтцфельдта-Якоба клинические и гистопатологические данные аналогичны таковым в спорадических случаях. В США только 10% случаев болезни Крейтцфельдта-Якоба относят к семейным. Синдром Герстмана-Штраусслера-Шейнкера и фатальная семейная бессонница — семейные формы заболевания. В некоторых затронутых семьях у 50% братьев, сестер и детей пациента с семейной формой болезни Крейтцфельдта-Якоба в конечном итоге развивается симптоматическое заболевание.
Ген, кодирующий PrP, тесно связан, если не идентичен гену, контролирующему периоды инкубации скрепи у овец и скрепи и болезни Крейтцфельдта-Якоба у мышей. Ген, кодирующий PrP у человека, называется геном PRNP и расположен на коротком плече хромосомы 20. Он имеет открытую рамку считывания из 759 нуклеотидов (253 кодона), в которой >20 различных точечных мутаций и множество встроенных последовательностей, кодирующих дополнительные тандемно-повторяющиеся октапептиды. Они обусловливают развитие губчатой энцефалопатии в семьях с паттерном, совместимым с аутосомным доминированием переменной пенетрантности.
Такая же нуклеотидная замена в кодоне 178 гена PRNP, связанного с болезнью Крейтцфельдта-Якоба, была обнаружена в некоторых семьях у всех пациентов с фатальной семейной бессонницей. Гомозиготность по валину (V) и особенно по метионину (М) по кодону 129 увеличивает восприимчивость к ятрогенной болезни Крейтцфельдта-Якоба и спорадической болезни Крейтцфельдта-Якоба. Почти все пациенты с новым вариантом болезни Крейтцфельдта-Якоба, подлежащие генотипированию, были гомозиготными по метионину по 129-му кодону гена PRNP. Несколько вероятных доклинических инфекций нового варианта болезни Крейтцфельдта-Якоба и 2 клинически типичных случая (один подтвержденный, а др. не полностью изученный) произошли у лиц с гетерозиготным генотипом 129 MV.
При секвенировании генов PRNP из приложений, содержащих PrPTSE в Великобритании, только 10% британских субъектов оказалось гомозиготным по V-генотипу. PrPTSE никогда не обнаруживался при новом варианте болезни Крейтцфельдта-Якоба. Значение этого открытия неизвестно. Власти Великобритании предположили, что в лимфоидных тканях некоторых людей с PrPTSE возможна скрытая персистенция инфекции. Неизвестно, заразна ли кровь таких людей.
У человека из семьи с болезнью Крейтцфельдта-Якоба или синдромом Герстмана-Штраусслера-Шейнкера и ассоциированными мутациями в PRNPgene выше вероятность развития симптоматической губчатой энцефалопатии. Носители ассоциированных с трансмиссивными губчатыми энцефалопатиями мутаций использовали доимплантационную генетическую диагностику и отбор эмбрионов in vitro, чтобы избежать передачи мутантного гена потомству.
Значение мутаций в генах PRNP у людей из семей, не страдающих губчатой энцефалопатией, неизвестно. Не следует беспокоить ЗЛ с разными мутациями в гене PRNP, поскольку последствия еще не ясны. В США люди не могут сдавать кровь, если кровному родственнику был поставлен диагноз трансмиссивной губчатой энцефалопатии. Исключение — случаи, когда у донора отсутствуют мутации, связанные с трансмиссивными губчатыми энцефалопатиями.
и) Прогноз. Прогноз всех губчатых энцефалопатий одинаково неблагоприятный. Ок. 10% пациентов могут выжить >1 года, но качество жизни значительно снижено.
к) Семейная поддержка. Фонд болезни Крейтцфельдта-Якоба, организованный и поддерживаемый членами семей и друзьями пациентов с болезнью Крейтцфельдта-Якоба и связанными с ней расстройствами, сотрудничает с CDC, с Национальным центром наблюдения за патологией прионных заболеваний. Университет Кейс Вестерн Резерв, Кливленд — группа поддержки и обучения для пациентов, а также полезный источник информации и доступных ресурсов для тех, кто изучает прионные болезни
л) Профилактика. Воздействие агента энцефалопатии крупного рогатого скота в мясных продуктах представляет особую опасность. Власти Канады, США и др. стран отреагировали на риск развития заболеваний введением в течение последних 20 лет более строгих мер в области сельского хозяйства и 30. Исключение большинства материалов крупного рогатого скота из кормов для животных — наиболее эффективная мера. В США с 2004 по 2012 гг. выявлено 3 случая энцефалопатии крупного рогатого скота у местных животных. Один случай обнаружен у канадской коровы, импортированной в США в 2003 г. С 2003 по 2015 г. Канада выявила 20 местных коров с энцефалопатией крупного рогатого скота (и завезла случай из Соединенного Королевства в 1993 г.).
Несмотря на обнадеживающие эпидемиологические исследования, которые не смогли выявить воздействие агентов скрепи или хронической истощающей болезни на трансмиссивные губчатые энцефалопатии человека, разумно ограничить контакт детей с мясом и др. продуктами, которые м.б. заражены каким-либо агентом.
Безопасность человеческой крови, компонентов крови и производных плазмы в США и Канаде защищена отсрочкой доноров с повышенным риском трансмиссивных губчатых энцефалопатий в анамнезе. К ним относят лиц, получавших трупные гормоны гипофиза (больше не применяются) или перенесших аллотрансплантацию ТМО, пациентов с семейным анамнезом болезни Крейтцфельдта-Якоба (если секвенирование не показывает, отсутствие мутации в генах PRNP у кровного родственника с трансмиссивной губчатой энцефалопатией или донора), и лиц, которые провели значительные периоды времени в определенных странах в те годы, когда энцефалопатия крупного рогатого скота была распространена.
Людям, которым переливали кровь в Соединенном Королевстве и Франции после 1980 г., следует отказать в сдаче крови (аналогичные правила отсрочки существуют для доноров человеческих клеток и тканей). Власти Великобритании предупредили лиц, получавших в период с 1989 по 2001 гг. объединенные концентраты факторов свертывания крови или антитромбин из Великобритании, что они м.б. «подвержены риску заражения болезнью Крейтцфельдта-Якоба» и что к ним применяются «особые меры инфекционного контроля».
Рекомендовано идентифицировать нескольких доноров крови и тканей, действительно инфицированных агентами трансмиссивных губчатых энцефалопатий, а не исключать доноров с повышенным риском заражения, т.к. большинство из них вряд ли были инфицированы. Прижизненные скрининговые тесты, которые могут со временем выявить доноров с доклиническими инфекциями, находятся в стадии разработки, хотя клинически не подтверждены. Маловероятно, что какой-то тест будет принят для скрининга доноров крови без одновременного проведения высокоспецифичного валидированного подтверждающего теста, чтобы избежать серьезных неблагоприятных последствий неизбежных л/п-результатов скрининга.
При обращении со всеми тканями, кровью и биологическими жидкостями человека следует соблюдать стандартные меры предосторожности. С материалами и поверхностями, загрязненными тканями или жидкостями пациентов с подозрением на болезнь Крейтцфельдта-Якоба, следует обращаться с большой осторожностью. По возможности выбрасывать загрязненные инструменты, тщательно упаковывая и сжигая. Загрязненные ткани и биологические продукты, вероятно, не м.б. полностью избавлены от инфекционных агентов без разрушения их структурной целостности и биологической активности. Именно поэтому следует изучать мед. и семейные истории отдельных доноров тканей, чтобы исключить трансмиссивные губчатые энцефалопатии.
Гистопатологическое исследование тканей ГМ трупных доноров и тестирование на аномальные PrP м.б. выполнены, чтобы обеспечить дополнительную гарантию безопасности. Хотя нельзя полагаться на какой-либо метод стерилизации для удаления всех агентов с загрязненных поверхностей, воздействие влажного тепла, гидроксида натрия, хлорного отбеливателя, концентрированной муравьиной кислоты, подкисленного детергента и солей гуанидина заметно снижало инфекционный потенциал в экспериментальных исследованиях.