Желто-белая пятнистость сетчатки при системных болезнях
а) Сетчатка при абеталипопротеинемии (синдроме Бассена-Корнцвейга). Абеталипопротеинемия — редкое аутосомно-рецессивное расстройство метаболизма, при котором нарушается абсорбция холестерина, жиров пищи и жирорастворимых витаминов из тонкой кишки. Состояние сопровождается такими системными изменениями, как мальабсорбция жиров, отсутствие беталипопротеина в сыворотке крови, аномальная форма эритроцитов (акантоцитоз) и атактическая нейропатия.
Изменения глаз при абеталипопротеинемии вариабельны, описаны разнообразные симптомы, в том числе офтальмоплегия, птоз, нистагм, анизокория, катаракта, ангиоидные полосы и прогрессирующая дистрофия сетчатки, характеризуемая появлением белых точек на уровне ПЭС и пигментными изменениями.
Это заболевание вызывается рецессивными мутациями гена МТТР, кодирующего микросомальный протеин — переносчик триглицеридов. Лечение витаминами А и Е замедляет течение заболевания и предотвращает развитие дегенерации сетчатки.
б) Сетчатка при синдроме Альпорта. Синдром Альпорта — генетически гетерогенное наследственное заболевание, которое может наследоваться по Х-сцепленному полудоминантному (85% случаев; ген COL4A5), аутосомно-рецессивному (15% случаев; гены COL4A3 и COL4A4) или аутосомно-доминантному (редко; ген COL4A3) типу. Это патология базальной мембраны, вызывающая прогрессирующую почечную недостаточность вследствие гломерулонефропатии, высокочастотную сенсоневральную тугоухость и различные аномалии глаз, в том числе катаракту, передний лентиконус, дистрофию роговицы и точечно-пятнистую ретинопатию.
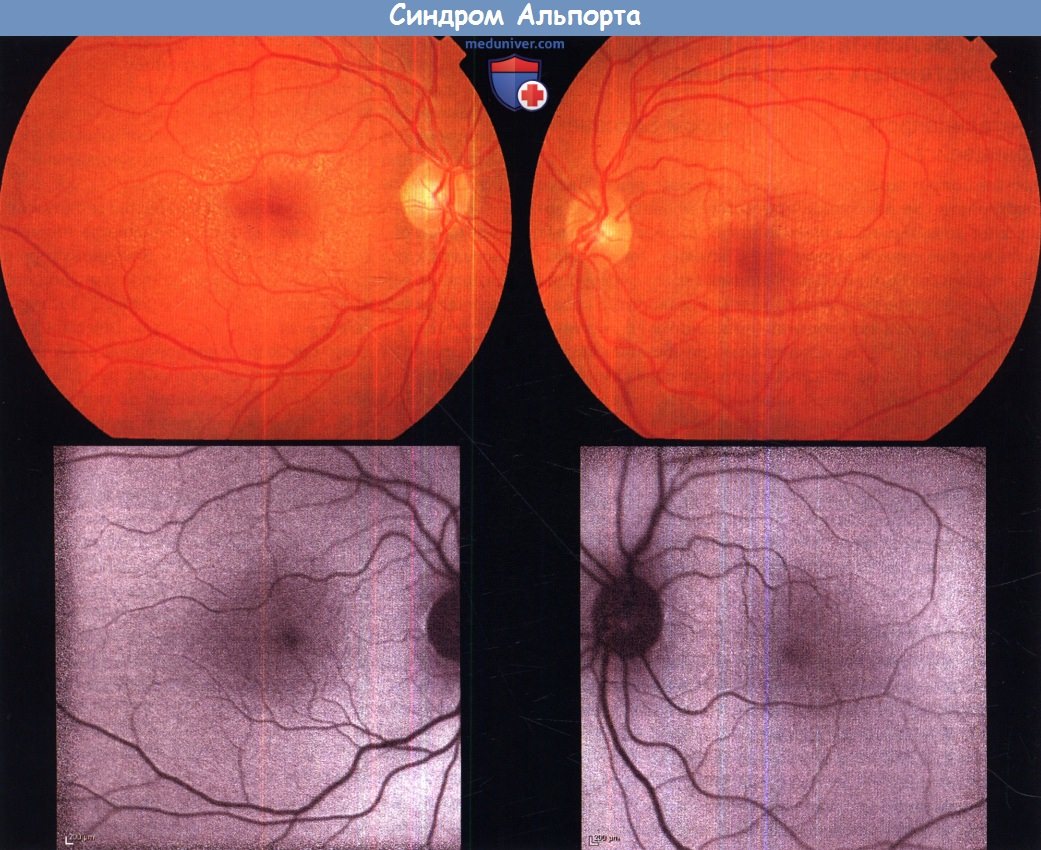
Поражение сетчатки обычно развивается у больных взрослых мужчин и представляет собою многочисленные желто-белые пятнышки перимакулярной области на уровне пигментного эпителия сетчатки; они отсутствуют в зоне центральной ямки, могут распространяться на периферию и могут напоминать точки при белоточечном глазном дне. Хотя и встречаются исключения, зрительные функции и результаты электрофизиологических исследований обычно в пределах нормы. Синдром Альпорта вызывается мутациями генов биосинтеза коллагена (COL4A3, COL4A4 и COL4A5), вызывающих дефекты синтеза или сборки сети коллагена IV типа.
Синдром Альпорта.
Фотография глазного дна левого и правого глаза 28-летнего пациента с синдромом Альпорта (вверху);
при исследовании флюоресценции (внизу) изменений не выявлено.
в) Сетчатка при эластической псевдоксантоме. Эластическая псевдоксантома (pseudoxanthoma elasticum — РХЕ) наследственное заболевание соединительной ткани, сопровождающееся характерными изменениями кожи (обвисшие складки относительно неэластичной кожи сгибательных поверхностей), а также поражением сетчатки и сердечно-сосудистой системы. У большинства больных идентифицированы рецессивные мутации гена АВСС6.
Тем не менее относительно часто встречается псевдодоминирование вследствие неожиданно высокой встречаемости в популяции носителей-гетерозигот, что необходимо учитывать при генетическом консультировании пациентов с эластической псевдоксантомой.
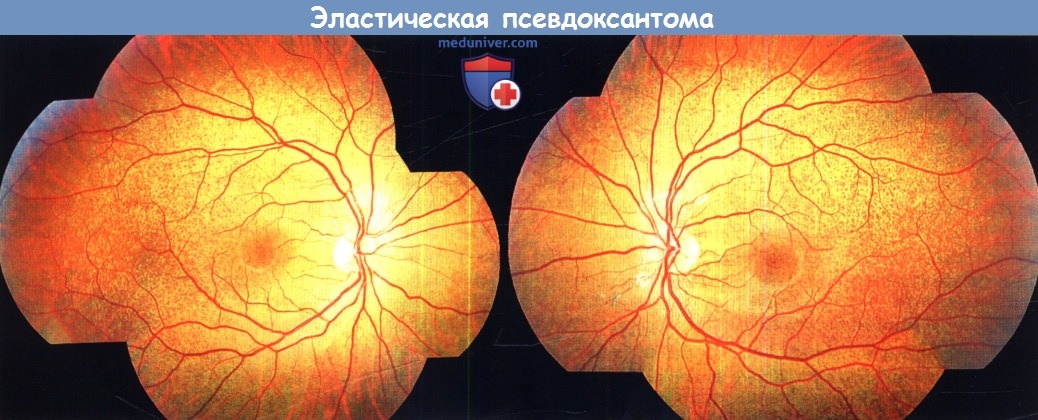
Наиболее часто встречающиеся изменения сетчатки при эластической псевдоксантоме — неоднородность пигментного эпителия сетчатки, первоначально с височной стороны от центральной ямки (апельсиновая корка) и ангиоидные полосы, тянущиеся от диска зрительного нерва. Последние встречаются почти у всех пациентов с эластической псевдоксантомой и часто приводят к хориоидальной неоваскуляризации, развитию фиброваскулярного рубца заднего полюса и снижению зрения. Изменения по типу апельсиновой корки предшествуют появлению ангиоидных полос и наблюдаются у маленьких пациентов в виде мелких желто-белых друзоподобных очагов в задне-височных отделах и на средней периферии глазного дна.
Исследование аутофлюоресценции очень информативно при обследовании пациентов с эластической псевдоксантомой, оно позволяет оценить протяженность поражения пигментного эпителия сетчатки. При ОКТ выявляются патологические изменения на уровне комплекса ПЭС/мембрана Бруха. Важное значение имеет раннее распознавание фенотипа эластической псевдоксантомы, который должен насторожить офтальмолога и ускорить направление пациента к дерматологу и кардиологу.
Эластическая псевдоксантома.
Фотография глазного дна левого и правого глаза 16-летнего больного.
г) Сетчатка при мембранопролиферативном гломерулонефрите II типа. Мембранопролиферативный гломерулонефрит II типа — редкая форма гломерулонефрита, характеризуемая наличием плотных отложений в пределах гломерулярной базальной мембраны. Отложения такого же состава и строения могут возникать у больных вдоль комплекса хориокапил-лярный слой — мембрана Бруха — пигментный эпителий сетчатки. Могут появляться друзоподобные отложения, часто наблюдаемые в области заднего полюса во втором десятилетии жизни. Их локализация варьирует у разных пациентов, по данным спектральной ОКТ они имеют другой состав и более разрушительны, чем друзы при возрастной макулярной дегенерации. Больные обычно не предъявляют жалоб, острота зрения остается хорошей.
Однако со временем по мере прогрессирования болезни, при помощи специализированных исследований, например ЭОГ и ЭРГ, могут выявляться нарушения функции сетчатки.
д) Сетчатка при первичной гипероксалурии. Первичная гипероксалурия — редкое врожденное нарушение метаболизма глиоксилата и оксалата, приводящее к повышению уровня оксалата в сыворотке крови и моче. При повышении уровня в сыворотке крови оксалат связывается с кальцием и образует нерастворимые кристаллы, которые откладываются в различных тканях, прежде всего в почках. Поражение почек, приводящее к хронической почечной недостаточности, является основной причиной преждевременной смерти.
Описано три различных типа заболевания. Наиболее часто встречающейся формой является гипероксалурия I типа, вызываемая недостаточностью печеночного лероксисомального фермента аланин-глиоксилат-аминотрансферазы (alanine: glyoxylate aminotransferase—AGT), сопровождающаяся аномалиями глаз. Первичная гипероксалурия обычно манифестирует в раннем детстве. У больных могут обнаруживаться множественные желтые кристаллические отложения на уровне пигментного эпителия сетчатки, более многочисленные в заднем полюсе и вдоль сосудистых аркад. Могут развиваться атрофические изменения макулы и появляться скопления пигмента. При исследовании аутофлюоресценции выявляются гипераутофлюоресцентные точки; при спектральной ОКТ видны гиперрефлективные образования ниже уровня пигментного эпителия сетчатки («куполообразная отслойка ПЭС»).
Отложения кристаллов оксалатов на сетчатке описаны у взрослых с вторичной гипероксалурией после анестезии метоксифлюраном.
Первичная гипероксалурия I типа.
Фотография глазного дна правого глаза больного.
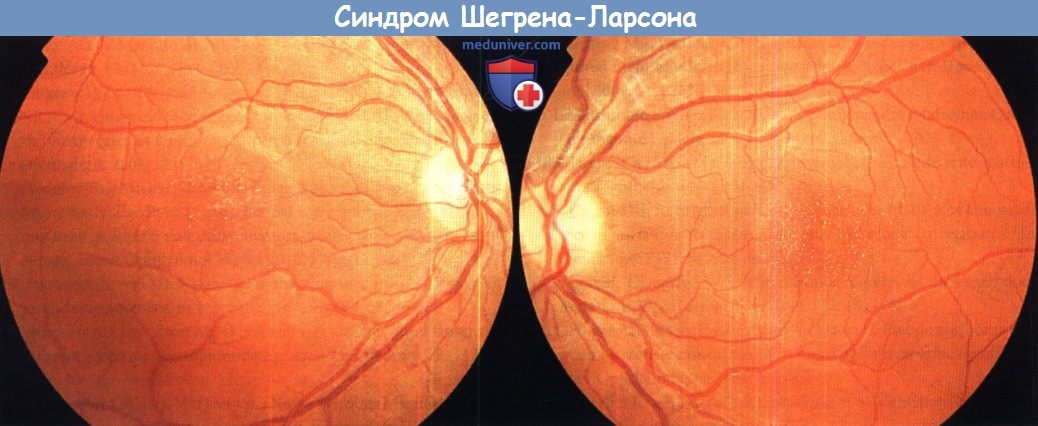
е) Сетчатка при синдроме Шегрена-Ларсона. Синдром Шегрена-Ларсона — наследуемое по аутосомно-рецессивному типу нейрокутанное заболевание детей, развивающееся вследствие недостаточности микросомального фермента дегидрогеназы жирных альдегидов (fatty aldehyde dehydrogenase — FALDH). Патологические изменения включают в себя врожденный ихтиоз, умственную отсталость и спастичность. Большинство больных рождается недоношенными, у них наблюдается характерная дебютирующая в детстве кристаллическая макулопатия, сопровождающаяся светобоязнью и ухудшением зрения. Описываются желто-белые блестящие отложения в макулярной области, возможны атрофические изменения пигментного эпителия сетчатки. Общая функция сетчатки сохранена.
ж) Различные сообщения о пятнистости сетчатки при синдромальном поражении глаз. Желто-белая пятнистость сетчатки может наблюдаться при нескольких системных заболеваниях, в том числе:
1. Синдром Kjellin (рецессивная мутация SPG11 или SPG15).
2. Синдром пятнистости сетчатки по типу «хлебных крошек».
3. Сенсоневральная тугоухость, аномалии пальцев и пятнистость сетчатки.
4. Синдром пятнистой сетчатки и кольцевой 17 хромосомы.
5. Недостаточность пероксисомального бифункционального ферментного комплекса.
Синдром Шегрена-Ларсона.
Фотография глазного дна левого и правого глаза 22-летнего больного.