Желто-белая пятнистость сетчатки при врожденной патологии глаз
Термином «пятнистая сетчатка» описываются расстройства, характеризуемые наличием множественных желто-белых очагов различных размеров и формы. Под это определение подпадают несколько приобретенных состояний, сопровождающихся ретинальной дисфункцией различной степени. В этой главе описана дифференциальная диагностика пятнистости сетчатки у детей; также описаны некоторые расстройства, встречающиеся у взрослых.
а) Клиническое обследование. Следует расспросить о жалобах со стороны зрения, таких как затуманивание зрения, никталопия или гемералопия, и составить полный семейный анамнез. Может быть информативным осмотр родственников, поскольку у других членов семьи заболевание может протекать бессимптомно. После этого необходимо расспросить о принимавшихся системных лекарственных препаратах и пищевых привычках. Отдельное внимание необходимо уделить другим имеющимся заболеваниям, особенно сопровождающимся мальабсорбцией (например, муковисцидоз).
При осмотре ребенка следует обращать внимание на распределение ретинальных отложений, глубину их залегания в сетчатке и являются ли они кристаллическими по структуре. Методы исследования включают в себя спектральную оптическую когерентную томографию, исследование аутофлюоресценции глазного дна (fundus autofluorescence imaging—FAF), и, в некоторых случаях, психофизическое и электрофизиологическое (электроретинография [ЭРГ] и электроокулографию [ЭОГ]) исследования. Для подтверждения диагноза может потребоваться проведение молекулярной диагностики.
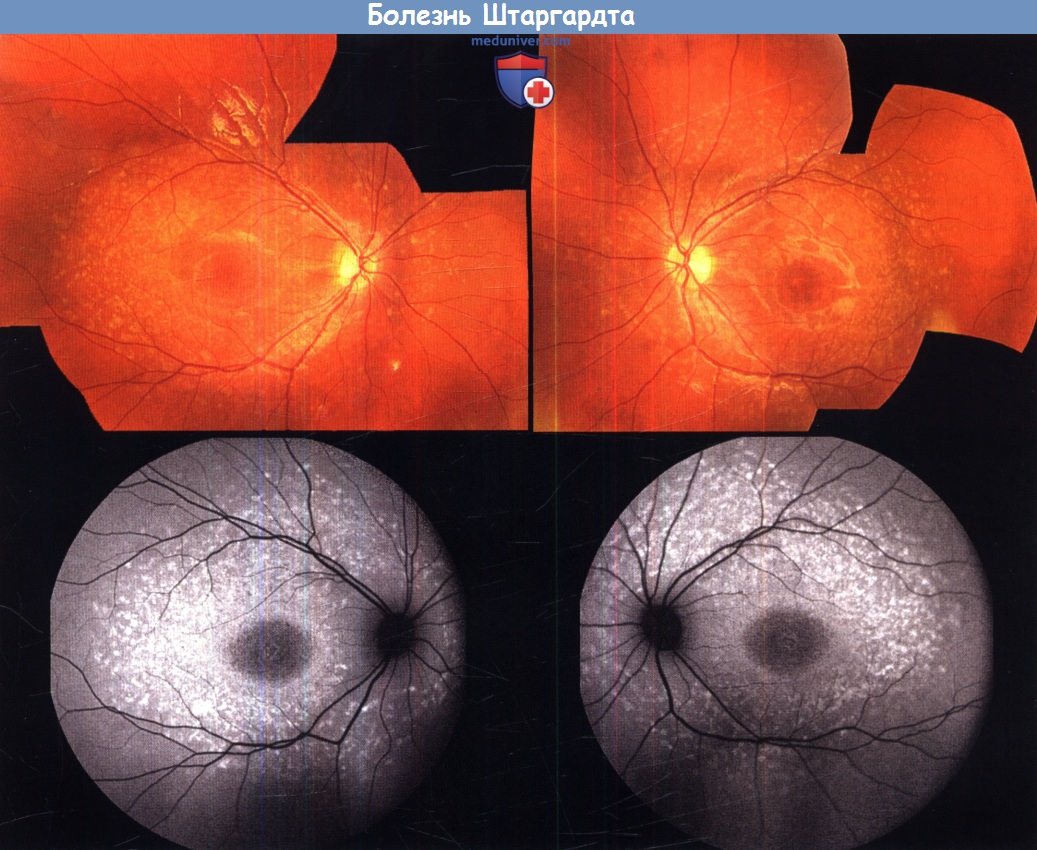
б) Болезнь Штаргардта и желтопятнистое глазное дно. Болезнь Штаргардта — дистрофия сетчатки, обычно наследуемая по аутосомно-рецессивному типу и вызываемая мутацией гена АВСА4. Псевдодоминантное наследование, когда у больного родителя (чей партнер, не зная об этом, является носителем заболевания) может родиться больной ребенок, может встречаться при близкородственных браках. В таких случаях большое значение для консультирования имеет дифференцировка АВСА4-связанного заболевания от фенотипически аналогичной аутосомно-доминантной патологии, вызванной мутацией ELOVL4 или RDS. Болезнь Штаргардта характеризуется желто-белой крапчатостью в области заднего полюса.
Часто развивается макулярная атрофия, при этом у больных обычно во втором десятилетии жизни снижается острота зрения. У некоторых пациентов при манифестации заболевания отмечается хорошее центральное зрение при минимальных или отсутствующих изменениях макулы, вследствие чего может быть диагностировано желтопятнистое глазное дно. Болезнь Штаргардта и желтопятнистое глазное дно составляют фенотипический спектр одного заболевания, и часто вызываются одними и теми же генетическими дефектами. При манифестации болезни пациенты редко жалуются на ухудшение ночного зрения и сужение полей зрения, хотя функция палочек может быть нарушена.
При диагностике болезни Штаргардта информативно исследование аутофлюоресценции; оно позволяет обнаружить патологический фенотип, даже если другими методами аномалии не выявляются. Даже на ранних стадиях заболевания при исследовании аутофлюоресценции выявляются изменения в форме крапчатого рисунка гипер- и гипоаутофлюоресценции и/или очагов усиления сигнала, соответствующих локализации пятен при офтальмоскопии. Данные спектральной ОКТ, а особенно степень сохранности линии, соответствующей границе между внутренними и наружными сегментами, являются более точным показателем тяжести заболевания. «Темная хориоидея» — классический признак заболевания при флюоресцентной ангиографии, хотя в настоящее время этот метод для диагностики болезни Штаргардта используется нечасто, особенно у детей.
При электрофизиологическом исследовании выявляется аномальная паттерн-ЭРГ и ослабление макулярных ответов центральной зоны макулы на мультифокальной ЭРГ. Результаты ганцфельд-электроретинографии вариабельны и часто на ранних стадиях остаются в пределах нормы. Ганцфельд-ЭРГ может быть прогностически информативной, поскольку у пациентов с ранними признаками дисфункции палочковых и/или колбочковых фоторецепторов отмечается более высокий риск тяжелого прогрессирующего заболевания.
Болезнь Штаргардта. Фотография глазного дна левого и правого глаза 9-летнего пациента с болезнью Штаргардта (верхний ряд).
Результаты исследования аутофлюоресценции (нижний ряд), видны гипераутофлюоресцирующие очаги, соответствующие пятнам сетчатки, и аномальная аутофлюоресценция зоны центральной ямки.
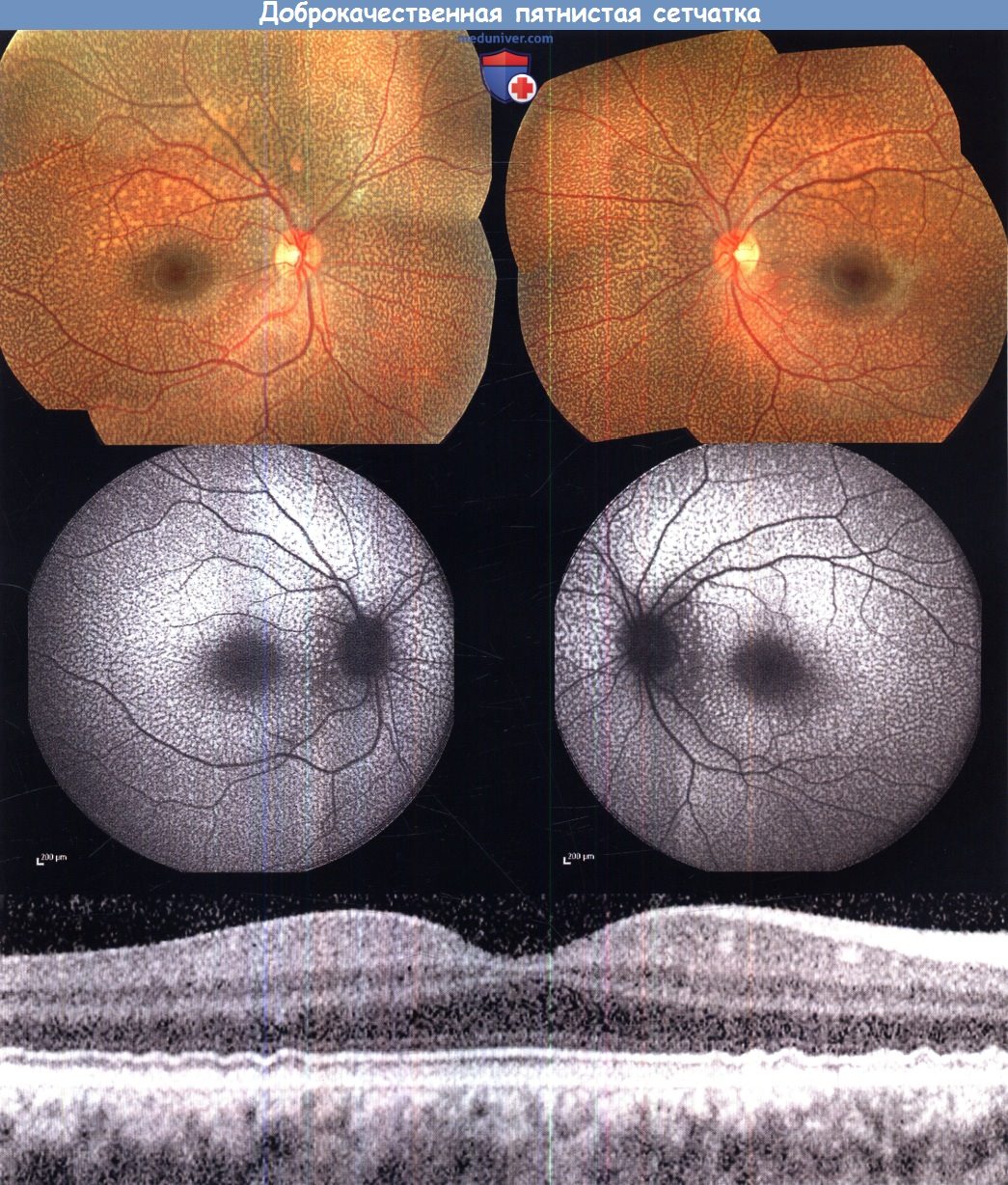
в) Доброкачественная пятнистая сетчатка. Доброкачественная пятнистая сетчатка — аутосомно-рецессивное состояние, характеризующееся характерной картиной изменений сетчатки и не сопровождающееся выраженными зрительными или электрофизиологическими нарушениями. Пациенты не предъявляют жалоб, но при офтальмоскопии выявляется яркая картина диффузных желто-белых пятнышек, распространяющихся до дальней периферии и отсутствующих в зоне центральной ямки. Пятнышки локализуются на уровне пигментного эпителия сетчатки (ПЭС) и могут выявляться уже в раннем младенческом возрасте.
При исследовании аутофлюоресценции выявляются множественные гипераутофлюоресцентные очаги, по локализации соответствующие пятнам на сетчатке. При спектральной ОКТ выявляются скопления четко отграниченных отложений, локализующихся кзади от линии соединения внутренних/наружных сегментов фоторецепторов, но не вызывающих ее деструкции. Диагноз подтверждается отсутствием изменений на ганцфельд- и паттерн-ЭРГ; на мультифокальной ЭРГ по некоторым изоптерам могут выявляться небольшие аномалии.
У некоторых пациентов с доброкачественной пятнистой сетчаткой были идентифицированы рецессивные мутации гена, кодирующего фосфолипазу А, V группы (PLA2G5) и слегка повышенные уровни липопротеинов низкой плотности и общего холестерина.
Доброкачественную пятнистую сетчатку не следует путать с «пятнистой сетчаткой Кандори». При этом состоянии на сетчатке наблюдаются крупные белые очаги, возможны атрофические изменения, и ночная слепота. Не ясно, является ли «пятнистая сетчатка Кандори» генетическим заболеванием и даже представляет ли собою отдельную нозологическую форму.
Доброкачественная пятнистая сетчатка. Фотография глазного дна левого и правого глаза 10-летнего пациента с доброкачественной пятнистой сетчаткой (верхний ряд).
Аутофлюоресценция глазного дна (средний ряд) и оптическая когерентная томография (горизонтальный центрированный по fovea линейный срез сетчатки правого глаза; внизу),
видны содержащие эндогенный флюорофор флюоресцирующие отложения на уровне пигментного эпителия сетчатки.
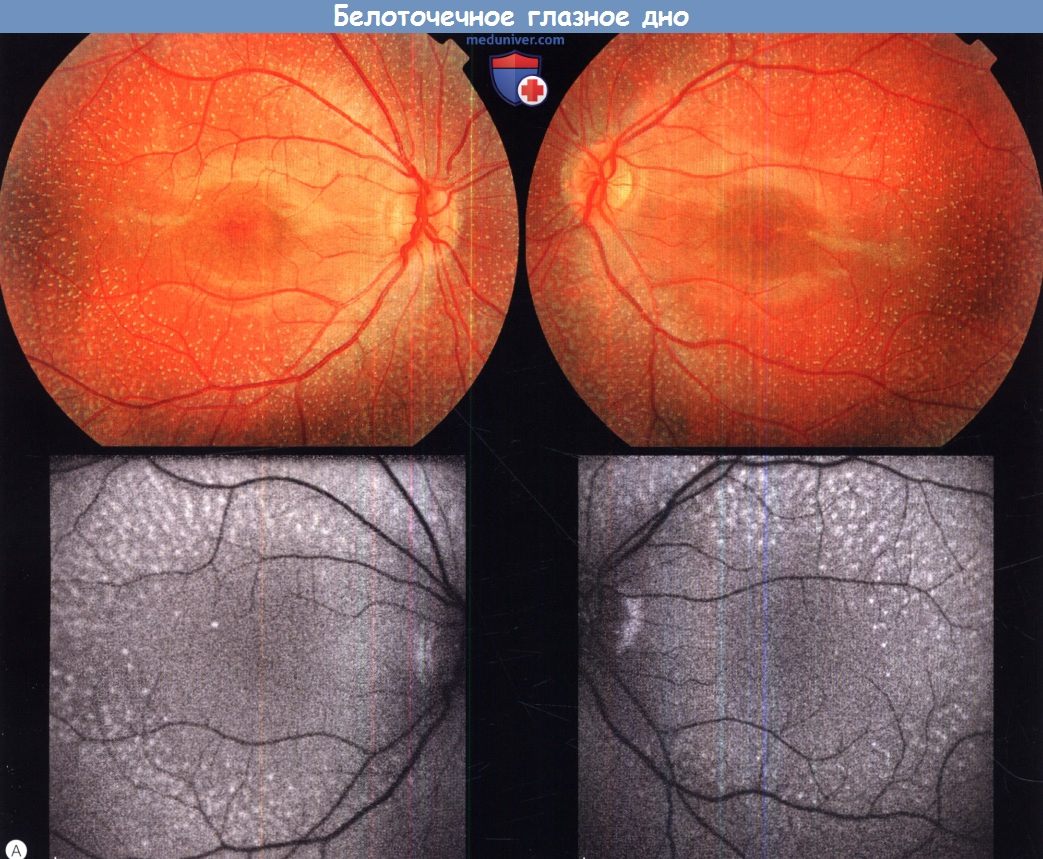
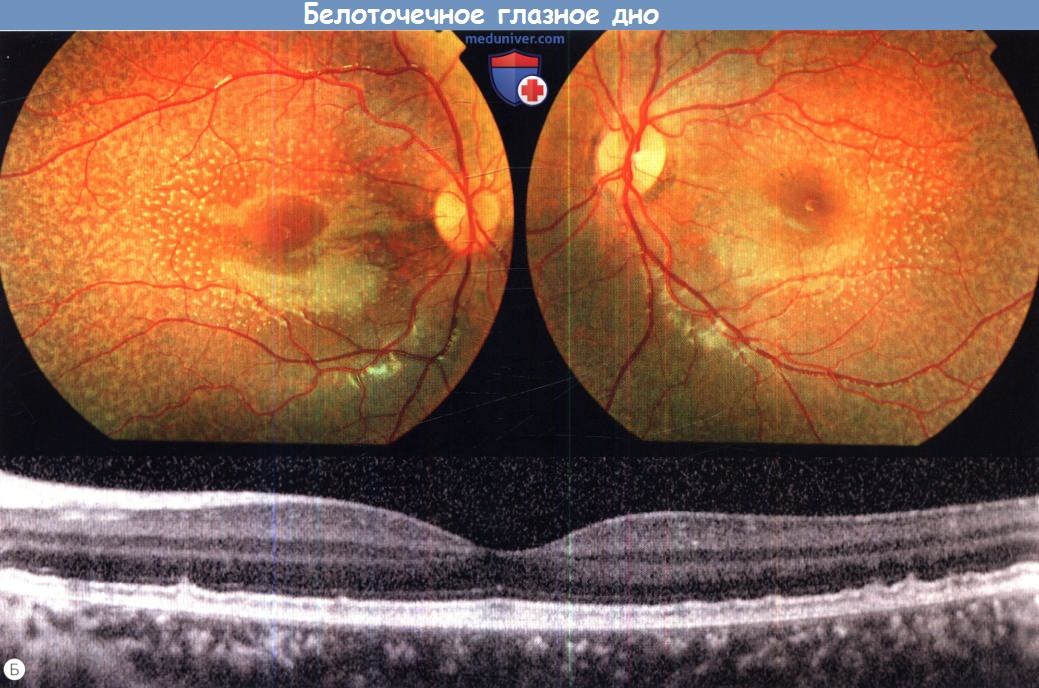
г) Белоточечное глазное дно (fundus albipunctatus). Белоточечное глазное дно — аутосомно-рецессивное состояние, сопровождающееся ночной слепотой и характеризующееся множественными широко распространенными желто-белыми точечными очагами на уровне пигментного эпителия сетчатки. Аномальная картина глазного дна выявляется при плановом обследовании или же пациент обращается за помощью по поводу никталопии. Больные жалуются на ночную слепоту с рождения и замедленную темновую адаптацию после воздействия яркого света. Острота зрения обычно в норме, поля зрения не изменены.
При исследовании аутофлюоресценции у пациентов с fundus albipunctatus часто отмечается снижение аутофлюоресценции; при цифровой обработке изображения могут визуализироваться артефакты, например аутофлюоресценция диска зрительного нерва и крупные сосуды. У маленьких детей могут выявляться очаги высокой плотности, частично соответствующие белым точкам, выявляемым при офтальмоскопии. При спектральной ОКТ выявляются гиперрефлективные очаги на уровне пигментного эпителия сетчатки.
Диагностике способствуют характерные аномалии, выявляемые при электрофизиологическом исследовании и адаптометрии. Примечательно, что при скотопической ЭРГ после стандартной темновой адаптации регистрируются субнормальные ответы, которые после длительной темновой адаптации нормализуются частично или полностью. Фотопическая ЭРГ и паттерн-ЭРГ могут быть изменены или нормальны.
Может быть информативным молекулярное исследование: белоточечное глазное дно обычно связано с рецессивными мутациями RDH5, гена, кодирующего фермент, обладающий 11-цис-ретинолдегидрогеназной активностью. Этот фермент в пигментном эпителии сетчатки катализирует окисление 11-цис-ретинол до 11-цис-ретинальдегида, универсального хромофора зрительных пигментов позвоночных. У пациентов с некоторыми признаками белоточечного глазного дна также были описаны мутации в генах RPE65 и RLBP1.
Белоточечное глазное дно.
(А) Фотография глазного дна левого и правого глаза 18-летнего пациента, результат анализа на мутацию RDH5 положительный (верхний ряд).
Исследование аутофлюоресценции глазного дна (нижний ряд), видны гипераутофлюоресцирующие очаги.
Белоточечное глазное дно.
(Б) Фотография глазного дна левого и правого глаза 10-летнего пациента, результат анализа на мутацию RDH5 положительный (верхний ряд).
При оптической когерентной томографии (горизонтальный центрированный по fovea линейный срез сетчатки правого глаза; внизу) определяются гиперрефлективные очаги на уровне пигментного эпителия сетчатки.
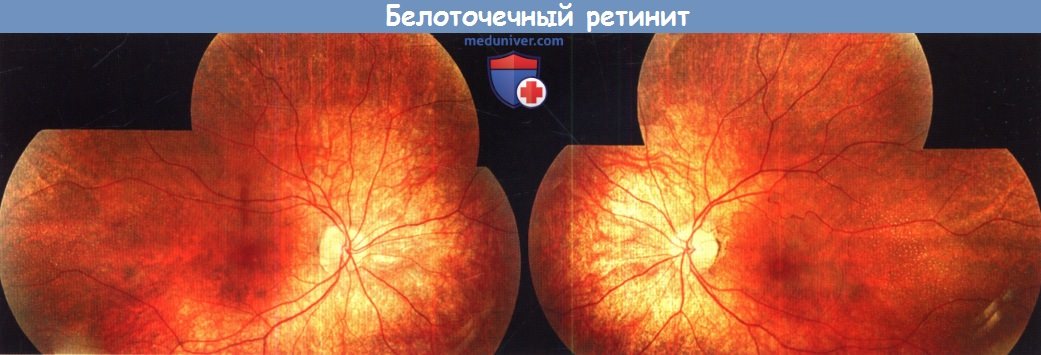
д) Белоточечный ретинит, дистрофии сетчатки Ботнического залива и Ньюфаундленда. Белоточечный ретинит (retinitits punctata albescens) — вариант пигментного ретинита, для которого характерно появление на сетчатке множества белых точек, а не отложений пигмента. Семейный анамнез таких пациентов свидетельствует об аутосомно-рецессивном типе наследования, заболевание манифестирует никталопией и/или нарушением периферического зрения; также может развиваться снижение остроты зрения. При офтальмоскопии выявляются белые точки, похожие на изменения при белоточечном глазном дне. Эти пятна отсутствуют в фовеолярной зоне, они могут эволюционировать с развитием более классической пигментной ретинопатии.
Важно дифференцировать начальные проявления белоточечного ретинита от белоточечного глазного дна: второе из этих двух состояний характеризуется значительно лучшим прогнозом для дневного (фотопического) зрения, тогда как первое представляет собою тяжелую прогрессирующую дегенерацию сетчатки. Ключевыми отличиями являются сужение сосудов, дефекты полей зрения и более тяжелые изменения при электрофизиологических исследованиях.
Белоточечный ретинит связан прежде всего с рецессивными мутациями гена ретиналь-связывающего протеина-1 (RLBP1). Также в редких случаях наследуемого по доминантному типу белоточечного ретинита описаны мутации генов RDS и родопсина.
Палочко-колбочковая дистрофия Ньюфаунленда и дистрофия Ботнического залива — две рано дебютирующие формы патологии сетчатки, часто встречающие в генетически изолированных популяциях соответственно северо-восточной Канады и северной Швеции. Оба заболевания имеют те же этиологию (мутации RLBP1) и основные фенотипические проявления (множественные белые точки, ночная слепота), что и рецессивный белоточечный ретинит и входят в фенотипический спектр этого заболевания.
Белоточечный ретинит.
Фотография глазного дна левого и правого глаза 15-летнего пациента, анализ на мутацию RLBP1 положительный.
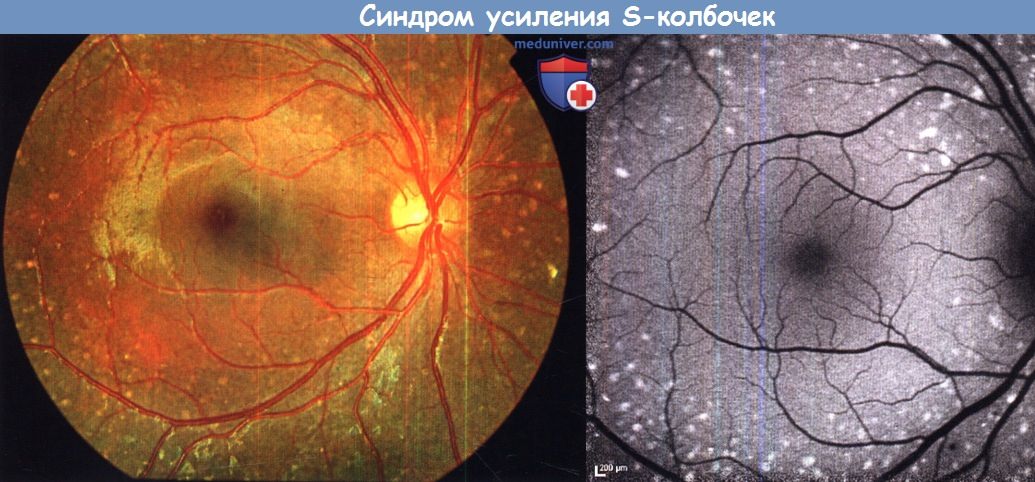
е) Синдром усиления S-колбочек и синдром Гольдмана-Фавра. Мелкие субретинальные белые точки могут являться ранним симптомом прогрессирующей ретинопатии называемой «синдромом усиления S-колбочек» («enhanced S-cone syndrome» — ESCS). Заболевание обычно манифестирует в первом или втором десятилетии жизни ночной слепотой и/или снижением остроты зрения; часто развивается дальнозоркость. Синдром усиления S-колбочек может сопровождаться различными изменениями глазного дна: наиболее типичные из них — ретиношизис и глубокие нумулярные пигментные отложения с желто-белыми пятнами или без них, первоначально появляющихся вдоль сосудистых аркад. У детей описаны белые точки, которые имеют тенденцию прогрессировать в пожилом возрасте в пигментную ретинопатию.
При исследовании аутофлюоресценции фиксируется аномальный сигнал, особенно к периферии от сосудистых аркад; у маленьких детей часто выявляются гипераутофлюоресцирующие пятна на средней периферии, лишь частично совпадающие с хлопьевидными аномалиями, видимыми при офтальмоскопии. Также описано гипераутофлюоресцентное кольцо. При ОКТ могут выявляться кистозные пространства, изменения типа розетки и/или ретиношизис.
Синдром усиления S-колбочек—уникальная дистрофия сетчатки: отмечается и дегенерация сетчатки, вызывающая ухудшение зрения, и усиление функции одного подтипа фоторецепторов — S-колбочек. Регистрируются характерные электрофизиологические изменения.
Синдром усиления S-колбочек вызывается рецессивной мутацией гена NR2E3, кодирующего ядерный рецептор сетчатки, участвующий в активации развития палочек и подавления развития колбочек.
Синдром Гольдмана-Фавра первоначально был описан как аутосомно-рецессивная витреоретинопатия, характеризуемая ночной слепотой, пигментной дегенерацией, макулярным и периферическим ретиношизисом, катарактой и дегенеративными изменениями стекловидного тела.
Многие из этих признаков общие с синдромом усиления S-колбочек; в результате соответствующих генетических и электрофизиологических исследований было показано, что эти два состояния являются не отдельными нозологиями, а двумя различными фенотипами широкого спектра клинической экспрессии одной дегенерации сетчатки.
Синдром усиления S-колбочек.
Фотография глазного дна и результат исследования аутофлюоресценции левого глаза 13-летнего пациента.
ж) Различные описания пятнистости сетчатки, связанной с наследственными заболеваниями сетчатки. Мелкие желто-белые хлопьевидные очаги описаны при различных других заболеваниях сетчатки, в том числе при рано дебютирующей дистрофии вследствие мутации RPE65 и ювенильным ретиношизисе.В первом случае точки обычно более мелкие и локализуются дальше на периферии; при втором заболевании они локализуются в области заднего полюса. Также аутосомно-рецессивная бестрофинопатия, состояние, развивающееся на фоне биаллельной мутации BEST1, может манифестировать в детстве появлением мелких желто-белых субретинальных отложений в зоне центральной ямки и вдоль сосудистых аркад.
з) Друзоподобные отложения при наследственных заболеваниях сетчатки. Появление друз в макулярной области является типичным признаком возрастной макулонатии. Такие же изменения могут наблюдаться у молодых людей с дистрофией глазного дна Сорсби (мутация гена TIMP3) и аутосомно-доминантными друзами, состоянием, также известным как сотовидная дистрофия сетчатки Дауна или malattia leventinesе (мутация гена EFEMP1) — двумя макулопатиями, наследуемыми по доминантному типу. Иногда друзоподобные изменения как случайная находка обнаруживаются у практически здоровых людей.
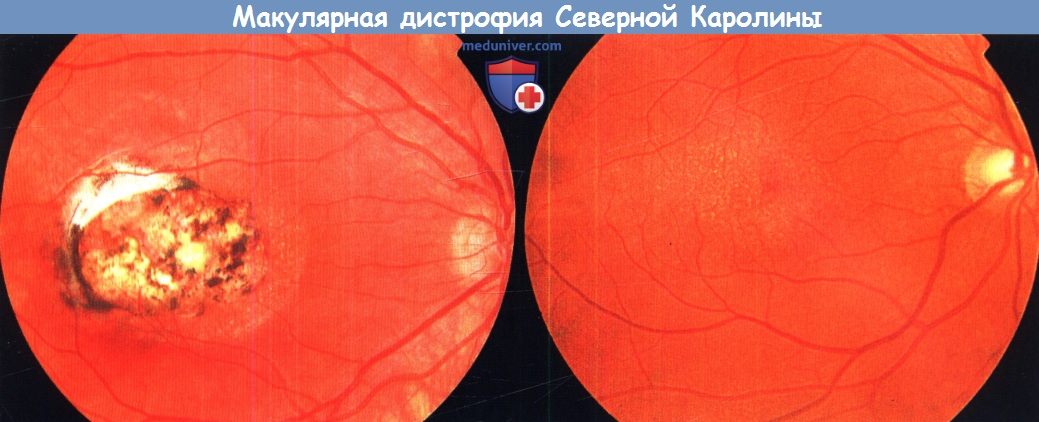
Друзоподобные отложения могут встречаться у детей с макулярной дистрофией Северной Каролины. Макулярная дистрофия Северной Каролины — аутосомно-доминантное состояние, характеризуемое выявляемыми с рождения друзами макулярной области. Офтальмоскопические изменения крайне вариабельны и могут быть гораздо более выраженными, чем можно было бы ожидать на основании визометрии. У больных могут наблюдаться:
1. Лишь несколько друз в зоне центральной ямки (I степень).
2. Сливные друзы с изменениями ПЭС или без таковых (II степень),
или
3. Центральная макулярная хориоретинальная атрофия с гипертрофией фиброзной ткани (III степень).
Прогресс заболевания наблюдается редко; приведенная классификация не отражает последовательные стадии развития патологических изменений. Острота зрения стабильна и в большинстве сохранна, за исключением случаев развития центральной атрофии. Хориоидальная неоваскуляризация развивается нечасто, но приводит к фиброзу и, позже, к слепоте.
Хотя вызывающий заболевание генетический дефект неизвестен, ответственный ген картирован на длинном плече шестой хромосомы. Описано несколько макулярных дистрофий, гены которых локализуются не в 6 хромосоме, но имеют общие признаки с макулярной дистрофией Северной Каролины (в том числе друзы макулярной области).
Макулярная дистрофия Северной Каролины.
Фотография глазного дна правого глаза 12-летнего мальчика (левая фотография) и его 40-летнего отца (правая фотография).
Оба страдают макулярной дистрофией Северной Каролины.
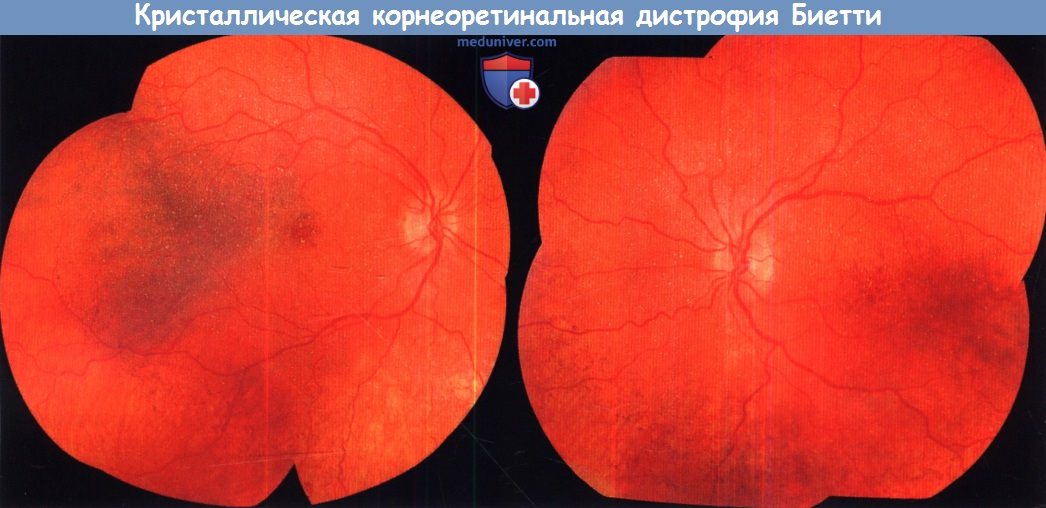
и) Кристаллическая корнеоретинальная дистрофия Биетти. Кристаллическая дистрофия Биетти — аутосомно-рецессивное заболевание, характеризующееся прогрессирующей хориоретинальной дегенерацией, никталопией, снижением остроты зрения, сужением полей зрения и наличием множественных желто-белых кристаллических отложений на сетчатке, в лимфоцитах крови и, иногда, в лимбе. Кристаллические отложения сетчатки выявляются преимущественно в области заднего полюса, в поверхностных и глубоких слоях сетчатки. В месте их локализации возникают множественные четко отграниченные зоны атрофии пигментного эпителия сетчатки; с развитием заболевания кристаллические отложения исчезают. Жалобы у больных обычно возникают на третьем десятилетии жизни.
Однако описано несколько случаев развития заболевания у детей. Изменения, по локализации частично соответствующие кристаллическим отложениям, могут выявляться при исследовании аутофлюоресценции, хотя и не всегда. При спектральной ОКТ выявляются гиперрефлективные образования по всей поверхности сетчатки; локализация многих этих элементов не соответствует локализации кристаллических отложений сетчатки, выявляемых при офтальмоскопии. При ганцфельд-ЭРГ даже на ранних стадиях заболевания выявляется поражение и палочковой, и колбочковой систем.
Корнеоретинальная дистрофия Биетти вызывается дефектами гена CYP4V2 (цитохром Р450, семейство 4, подсемейство V, полипептид 2), кодирующего фермент, участвующий в метаболизме жирных кислот. Высокая встречаемость носителей-гетерозигот мутации CYP4V2, и, следовательно, относительно высокая заболеваемость этой патологией отмечена в Восточной Азии, особенно среди населения Китая и Японии.
Описана наследуемая по доминантному типу ретинопатия (с неполной пенетрантностью и вариабельной экспрессивностью) с такими же клиническими проявлениями, что наблюдаются при кристаллической дистрофии Биетти.
Кристаллическая корнеоретинальная дистрофия Биетти.
Фотография глазного дна левого и правого глаза 16-летнего пациента, результат анализа на мутацию CYP4V2 положительный,
у больного наблюдаются кристаллические отложения.