Генетика семейной гиперхолестеринемии. Наследование, молекулярные основы
Открытие класса болезней, вызванных дефектами в молекулах рецепторов, произошло после идентификации в 1974 г. Голдстейном и Брауном рецептора ЛПНП как полипептида, повреждаемого при наиболее частой форме семейной гиперхоле-стеринемии. Это заболевание, приводящее к значительному повышению риска инфаркта миокарда, характеризуется повышением холестерина плазмы, переносимого ЛПНП, главным транспортным белком холестерина в плазме.
Открытие Голдстейна и Брауна в общих чертах прояснило нормальный метаболизм холестерина и биологию поверхностных рецепторов клетки. Недостаточность рецептора ЛПНП — первый пример из множества выявленных впоследствии нарушений, вызванных дефектами рецепторов.
Семейная гиперхолестеринемия относится к группе метаболических нарушений, получивших название гиперлипопротеинемий, характеризующихся высокими уровнями липидов (холестерина, триглицеридов, или и того и другого сразу) и специфических липопротеинов плазмы. Описаны также и другие моногенные гиперлипопротеинемий, каждая с четким биохимическим и клиническим фенотипом.
Оказалось, что, кроме мутаций в гене рецептора ЛПНП, к семейной гиперхолестеринемии также ведут аномалии в трех других генах. Примечательно, что все четыре гена, ответственные за семейную гиперхолестеринемию, нарушают либо функцию или количество рецептора ЛПНП в правильном положении на поверхности клетки, либо апопротеин В-100, белковый компонент лиганда ЛПНП-рецептора. Неудивительно, что клинические фенотипы индивидуумов, несущих мутации в этих четырех генах, трудноразличимы. Вследствие ее значимости рассмотрим семейную гиперхолестеринемию, вызванную мутациями в рецепторе ЛПНП.
Мы обсудим также мутации в гене протеазы PCSK9; хотя некоторые мутации в этом гене вызывают гиперхолестеринемию, большее значение PCSK9 в том, что ее некоторые частые варианты понижают уровень ЛПНП-холестерина плазмы в общей популяции, защищая от заболевания коронарных сосудов сердца.
Семейная гиперхолестеринемия вследствие мутаций в рецепторе ЛПНП
Мутации в гене, кодирующем рецептор ЛПНП, — самая частая причина семейной гиперхолестеринемии. Рецептор представляет собой поверхностный клеточный белок, ответственный за связывание ЛПНП и доставку его внутрь клетки. Как у гетерозигот, так и у гомозигот развивается ранняя патология сердца в результате появления атером (отложения производных холестерина и ЛПНП в венечных артериях), ксантом (холестерин депонируется в коже и сухожилиях) и старческой дуги (отложение холестерина вокруг периферии роговицы).
Так тщательно пока изучены немногие болезни; полностью подтверждена вся последовательность патологических событий от мутантного локуса до его эффекта на уровне человека и популяции.
Генетика семейной гиперхолестеринемии вследствие мутаций в рецепторе ЛПНП. Семейная гиперхолестеринемия, вызванная мутациями в рецепторе ЛПНП, наследуется как аутосомный полудоминирующий признак. Известны как гомозиготные, так и гетерозиготные фенотипы, очевиден эффект дозы генов; болезнь обнаруживается раньше и проявляется сильнее у гомозигот, чем у гетерозигот, отражая большее снижение числа рецепторов ЛПНП и большее повышение ЛПНП в плазме.
Гомозиготы могут уже в детстве иметь клинически значимое заболевание коронарных сосудов сердца, немногие доживают до 40 лет. Гетерозиготная форма болезни с популяционной частотой около 1 на 500 — одно из наиболее частых моногенных заболеваний человека. Гетерозиготы имеют высокие уровни холестерина плазмы, почти в 2 раза выше, чем в контрольной группе. По причине наследственной природы семейной гиперхолестеринемии важно подтвердить или отвергнуть этот диагноз, встречающийся приблизительно у 5% выживших после инфаркта миокарда гетерозигот по дефекту рецептора ЛПНП. В то же время семейную гиперхолестеринемию имеет только около 1 из 20 в общей популяции лиц с повышенным холестерином и гиперлипопротеинемией, аналогичной имеющейся при гетерозиготной недостаточности рецептора ЛПНП, чаще такие индивидуумы имеют гиперхолестеринемию многофакторного происхождения.
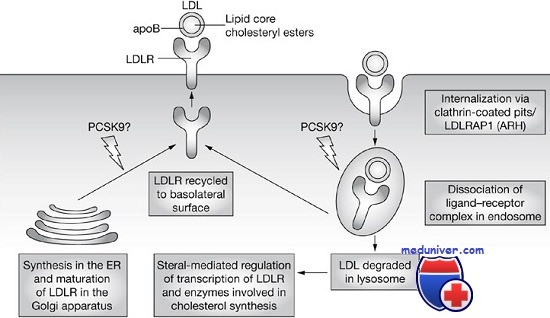
Захват холестерина рецептором ЛПНП. Нормальные клетки получают холестерин, или синтезируя его, или захватывая из плазмы внешний холестерин, связанный с ЛПНП. Захват осуществляет рецептор ЛПНП, распознающий апопротеин В-100, белковый остаток ЛПНП. Рецепторы ЛПНП на поверхности клетки локализуются в углублениях (закрытых ямках), создаваемых белком клатрином. Связанный рецептором ЛПНП переносится в клетку за счет эндоцитоза ямок, которые в конце концов попадают в лизосомы, где ЛПНП гидролизуются, освобождая холестерин. Увеличение свободного внутриклеточного холестерина уменьшает внутренний синтез холестерина, подавляя ограничивающий фермент пути синтеза (3-гидрокси-З-метилглутарил-коА-редуктазу). Холестерин, ненужный для клеточного метаболизма или синтеза мембран, может реэтерифицироваться и накапливаться в виде эфиров холестерина. Этот процесс стимулируется активацией ацетилкоэнзим-А-холестерол-ацилтрансферазы. Увеличение внутриклеточного холестерина также уменьшает синтез рецептора.
Классы мутаций в рецепторе ЛПНП. В гене рецептора ЛПНП обнаружено более 700 различных мутаций, распределенных по всей его длине. (Часто остается неясным, все ли эти варианты нуклеотидной последовательности патогенны или некоторые из них — варианты нормы без фенотипического проявления.) Подавляющее большинство аллелей — однонуклеотидные замены, небольшие инсерции или делеции; структурные перегруппировки занимают в большинстве популяций всего от 2-10% аллелей рецептора ЛПНП.
Зрелый рецептор ЛПНП имеет пять четких структурных областей, в основном имеющих различающиеся функции. В определении функций этих областей заметную роль сыграл анализ эффекта мутаций в каждой области рецептора. Эти исследования демонстрируют тот вклад, который может внести генетический анализ в определение связей между структурой и функциями белков.
Для исследования мутантных рецепторов и получающихся сбоев в клеточном метаболизме холестерина проведены исследования в культурах фибробластов больных. Мутации в гене рецептора ЛПНП можно сгруппировать в шесть классов, в зависимости от нарушаемого мутацией этапа нормального клеточного цикла рецептора. Мутации 1-го класса — аллели, полностью нарушающие синтез рецептора, наиболее частый тип патогенных мутаций в этом локусе. В остальных пяти классах рецептор синтезируется нормально, но нарушены его функции.
Мутации в классах 2, 4 и 6 определяют характеристики полипептида, критичные для его субклеточной локализации. Сравнительно частые мутации 2-го класса нарушают перенос рецепторов ЛПНП, и они накапливаются в месте их синтеза — эндоплазматическом ретикулуме вместо перемещения в комплекс Гольджи. Эти аллели нарушают соответствующую третичную структуру белка, необходимую для его выхода из эндоплазматического ретикулума. Мутантные рецепторы 3-го класса достигают поверхности клетки, но не способны связывать ЛПНП.
Таким образом, эти аллели позволили исследователям идентифицировать область связывания ЛПНП. Мутации 4-го класса нарушают прикрепление рецептора в клатриновой ямке и связанный ЛПНП не переносится в клетку. Данные мутации изменяют или удаляют цитоплазматическую область в карбоксильном конце рецептора, доказывая, что этот участок в норме направляет рецептор в ямку. Мутации 5-го класса — аллели, нарушающие восстановление рецептора. Для восстановления рецептора необходима его диссоциация со связанным ЛПНП в эндосоме. Мутации в гомологичном участке предшественника фактора роста эпидермиса нарушают освобождение лиганда ЛПНП. Это приводит к распаду рецептора, возможно, вследствие того, что занятый рецептор не может вернуться на поверхность клетки.
Протеазы PCSK9 и их отношения с ЛПНП и холестерином при гиперхолестеринемии
Миссенс-мутации с увеличением функции в гене, кодирующем протеазу PCSK9, оказались причиной редкой аутосомно-доминантной семейной гиперхолестеринемии. Эксперименты показывают, что повышенная активность протеазы PCSK9 ведет к распаду рецептора ЛПНП (хотя неясно, является ли рецептор ее прямой мишенью), регулируя тем самым уровень рецептора в гепатоцитах. Следовательно, протеаза действует как механизм, уменьшающий количество молекул рецептора и предохраняющий от чрезмерного захвата холестерина, что необходимо, например, в случае низкохолестериновой диеты.
Миссенс-мутации в протеазах PCSK9, связанных семейной гиперхолестеринемией, оказывается, вызывают болезнь, увеличивая активность протеаз, тем самым снижая содержание рецептора ЛПНП до аномально низкого уровня. Мутации в гене PCSK9 с увеличением функции, вызывающие семейную гиперхолестеринемию, показали, что протеазы PCSK9 — основной регулятор метаболизма холестерина ЛПНП.
Защитное действие некоторых вариантов нуклеотидной последовательности гена PCSK9 от ишемической болезни сердца
Связь между семейной гиперхолестеринемией и геном PCSK9 позволяет предполагать, что частые варианты нуклеотидной последовательности в гене PCSK9 могут быть связаны как с очень высоким, так и очень низким уровнем холестерина ЛПНП в общей популяции (несмотря на то что, к сожалению, для частых вариантов в других генах, включая три других, связанных с семейной гиперхолестеринемией, связь с изменениями уровня холестерина плазмы в общей популяции убедительно не доказана). Обнаружено несколько вариантов последовательности гена PCSK9, тесно связанных с низким уровнем холестерина ЛПНП в плазме.
Например, в афроамериканской популяции с очень низким уровнем холестерина ЛПНП один из двух нонсенс-вариантов PCSK9 обнаруживается в 2,6% всех случаев; присутствие любого варианта приводит к уменьшению уровня холестерина ЛПНП на 40%.
Это снижение холестерина ЛПНП оказывает мощный защитный эффект против ИБС, уменьшающий ее риск почти на 90%; только около 1% чернокожих носителей любого из двух вариантов нонсенс-мутаций за 15-летний период наблюдения имели ИБС, по сравнению с почти 10% без любой из этих двух мутаций. Другой аллель (Arg46Leu) — более частый (3,2%) у европеоидов и вызывает только 15% уменьшение уровня холестерина ЛПНП, но, что удивительно, на 50% уменьшает заболеваемость ИБС.
Эти сведения имеют большое значение для здравоохранения, поскольку они позволяют предполагать, что незначительное, но продолжающееся всю жизнь уменьшение холестерина ЛПНП плазмы на 20-40 мг% должно значительно уменьшить встречаемость ИБС в популяции. Наконец, эти открытия показывают, как исследование редкого генетического нарушения может привести к важному новому знанию о генетическом вкладе в частые генетически комплексные болезни.
Патогенез атеросклеротических бляшек при семейной гиперхолестеринемии
Несмотря на более чем 30-летний период исследований, приведший к впечатляющему расширению знаний биологии рецептора ЛПНП и семейства молекулярных дефектов, влияющих на развитие семейной гиперхолестеринемии, механизмы, которыми повышение уровня ЛПНП приводит к образованию атеросклеротических бляшек в артериях, все еще не ясны.
У гомозигот повышенный уровень ЛПНП снижается во внеклеточной жидкости альтернативными рецепторами — «падалыциками» некоторых клеток, например макрофагов. Исследования макрофагов in vitro показывают, что избыточный холестерин хранится в виде микрокапель эфиров холестерина, вызывая появление пенистых клеток, обычно обнаруживаемых в ксантомах и атеросклеротических бляшках, но значимость этого процесса in vivo в настоящее время не определена.
Наконец, выяснение биохимической основы семейной гиперхолестеринемии оказало глубокое влияние на лечение значительно более частых форм спорадической гиперхолестеринемии, приведя к созданию статинового класса лекарств, тормозящих биосинтез холестерина.