Генетика болезни Альцгеймера. Наследование, молекулярные основы
До недавних пор биохимические механизмы, лежащие в основе почти всех нейродегенеративных болезней с началом во взрослом возрасте, были полностью неясными. Одно из наиболее частых таких заболеваний — болезнь Альцгеймера. Болезнь Альцгеймера обычно проявляется на шестом-девятом десятке лет, но есть моногенные формы, часто дебютирующие раньше, иногда даже на третьем десятилетии жизни.
Клинические проявления болезни Альцгеймера характеризуются прогрессирующим ухудшением памяти и высших корковых функций, например аргументации, а также поведенческими изменениями. Эти аномалии отражают вырождение нейронов в специфических областях коры мозга и гиппокампе.
Болезнь Альцгеймера поражает около 1,4% лиц в развитых странах и вызывает за год только в Соединенных Штатах 100 000 смертей.
Генетика болезни Альцгеймера
Родственники пациентов с болезнью Альцгеймера первой степени родства имеют 38% риска развития болезни к 85-летнему возрасту. Следовательно, оказывается, что большинство случаев с семейным накоплением имеет сложный генетический вклад. Этот вклад может создаваться одним или более независимо действующих неполно пенетрантных генов, несколькими взаимодействующими генами или некоторой комбинацией генетических и средовых факторов.
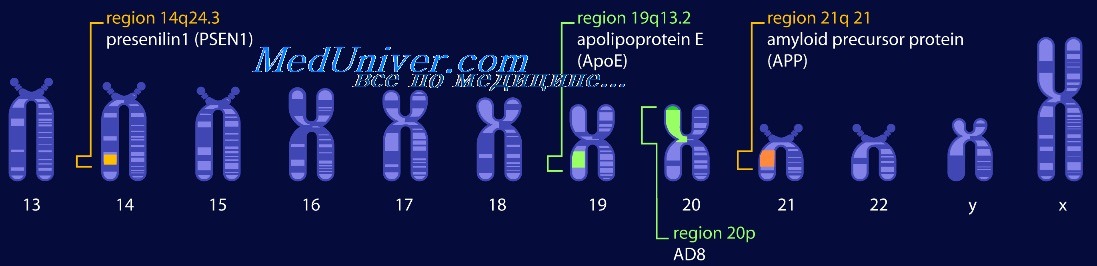
От 7 до 10% пациентов имеют моногенную высокопенетрантную форму болезни Альцгеймера, наследуемую по аутосомно-доминантному типу. В 1990-е годы обнаружено четыре гена, связанных с болезнью Альцгеймера. Мутации в трех из них, кодирующих бета-АРР, пресенилин 1 и пресенилин 2, ведут к аутосомно-доминантной болезни Альцгеймера. Четвертый ген, АРОЕ, кодирует АроЕ, белковый компонент нескольких плазменных липопротеинов.
Мутации в АРОЕ не связаны с моногенными формами болезни Альцгеймера. Аллель е4 АРОЕ несколько увеличивает восприимчивость к несемейной болезни Альцгеймера и влияет на возраст начала, по крайней мере, при некоторых моногенных формах.
Идентификация четырех генов, связанных с болезнью Альцгеймера, позволила проникнуть не только в патогенез моногенных форм болезни Альцгеймера, но так же, как это часто бывает в медицинской генетике, в механизмы, лежащие в основе более частой формы несемейной или «спорадической» болезни Альцгеймера. На самом деле, в центре патогенеза болезни Альцгеймера оказался избыток одного продукта протеолиза бета-АРР, названного А-бета-пептидом, и в настоящее время экспериментально подтверждено, что белки бета-АРР, пресенилин 1 и 2 вместе непосредственно участвуют в патогенезе болезни Альцгеймера.
Патогенез болезни Альцгеймера: бета-амилоидный пептид и отложение тау-белка
Наиболее важные патологические аномалии при болезни Альцгеймера — накопление в мозге двух фибриллярных белков, А-бета и белка тау. Пептид А-бета образуется из большего белка бета-АРР и обнаруживается во внеклеточном амилоиде или сенильных бляшках во внеклеточном пространстве мозга при болезни Альцгеймера.
Амилоидные бляшки, кроме пептида А-бета, содержат и другие белки, особенно АроЕ. Тау-белок — микротубулярный, обильно экспрессирующий в нейронах мозга. Гиперфосфорилированные формы тау-белка формируют нейрофибриллярные клубки, обнаруживаемые при болезни Альцгеймера, в отличие от внеклеточных амилоидных бляшек, внутри нейронов.
Тау-белок в норме обеспечивает сборку и устойчивость микротрубочек, эта функция уменьшается при фосфорилировании. Хотя образование клубков нейрофибрилл оказалось одной из причин гибели нейронов при болезни Альцгеймера, мутации в гене тау-белка связаны не с болезнью Альцгеймера, а с другим аутосомно-доминантным заболеванием, лобновисочной деменцией.
Бета-АРР — трансмембранный белок, подвергающийся трем различным видам протеолиза, в зависимости от относительной активности трех разных протеаз: а- и бета-секретаз — поверхностных клеточных протеаз; и у-секретазы — атипичной протеазы, расщепляющей мембранные белки в трансмембранных областях. Преобладающая судьба приблизительно 90% бета-АРР — расщепление а-секретазой, что предотвращает образование А-бета-пептида, так как а-секретаза расщепляет белок внутри него.
Оставшиеся приблизительно 10% бета-АРР расщепляются бета- и у-секретазами, формируя или нетоксичный пептид А-бета-40, или пептид А-бета-42, обладающий нейротоксичностью. Пептид А-бета-42 считают нейротоксичным, поскольку он более склонен к формированию нейрофибрилл, чем его аналог А-бета-40, признак, делающий болезнь Альцгеймера конформационной болезнью, подобно недостаточности а1-антитрипсина.
В норме образуется небольшое количество пептида А-бета-42; факторы, определяющие, будет ли белок расщепляться у-секретазой с образованием А-бета-40 или А-бета-42, не определены. При моногенной болезни Альцгеймера вследствие миссенс-замен в гене, кодирующем бета-АРР, тем не менее, несколько мутаций в гене бета-АРР избирательно увеличивают образование пептида А-бета-42. Это увеличение приводит к накоплению нейротоксичного А-бета-42 — основе патогенеза всех форм болезни Алыдгеймера, как моногенных так и спорадических.
Эта модель подтверждается тем, что пациенты с синдрома Дауна, имеющие три копии гена бета-АРР (расположенного в хромосоме 21), обычно имеют нейропатологические изменения болезни Альцгеймера уже в 40-летнем возрасте. Кроме того, мутации в генах пресенилина 1 и 2 также ведут к повышенному образованию А-бета-42. Примечательно, что в сыворотке больных с мутациями в генах бета-АРР, пресенилина 1 и 2 количество нейротоксичного пептида А-бета-42 повышается, и в культивируемых клетках экспрессия мутантных генов бета-АРР, пресенилина 1 и 2 увеличивает относительное образование пептида А-бета-42 в 2-10 раз.
Гены пресенилина 1 и 2 болезни Альцгеймера
Гены, кодирующие пресенилин 1 и пресенилин 2, обнаружены стратегией позиционного клонирования в семьях с аутосомно-доминантной формой болезни Альцгеймера. Пресенилин 1 необходим для расщепления у-секретазой производных бета-АРР. На самом деле, существуют подтверждения того, что пресенилин 1 — важный белковый кофактор у-секретазы.
Мутации в пресенилине 1 связаны с болезнью Альцгеймера через до сих пор неясный механизм, увеличивающий образование пептида А-бета-42. Белок пресенилин 2 имеет на 60% идентичную последовательность аминокислот с пресенилином 1, что указывает на их общие функции. Основное различие между мутациями в гене пресенилина 1 и 2 в том, что возраст начала во втором случае более вариабелен (пресенилин 1 — от 35 до 60 лет; пресенилин 2 — от 40 до 85 лет), в одной семье бессимптомный восьмидесятилетний носитель мутации в гене пресенилина 2 передал болезнь своему потомству. Эта разница частично зависит от числа е4 аллелей АРОЕ у носителей мутации в гене пресенилина 2; два е4 аллеля приводят к более раннему возрасту начала, чем один аллель, также обусловливающий более раннее начало по сравнению с другими аллелями АРОЕ.
Ген АРОЕ — локус восприимчивости к болезни Альцгеймера
Один аллель гена АРОЕ, е4 аллель, — основной фактор риска развития болезни Альцгеймера. Роль АРОЕ как основного локуса восприимчивости к болезни Альцгеймера была доказана четырьмя независимыми способами: анализом сцепления в семьях с накоплением болезни Альцгеймера с поздним началом, сильной ассоциацией аллеля е4 с болезнью Альцгеймера по сравнению с группой контроля, открытием того, что белок АроЕ — компонент амилоидных бляшек при болезни Альцгеймера, и обнаружением факта, что АроЕ связан с пептидом А-бета.
Белок АроЕ имеет три частых формы, кодируемые соответствующими аллелями АРОЕ. Аллель е4 значительно преобладает среди пациентов с болезнью Альцгеймера (40% по сравнению с 15% в общей популяции) и связан с ранним началом болезни (для гомозигот по аллелю е4 возраст начала болезни Альцгеймера на 10-15 лет меньше, чем в общей популяции). Кроме того, отношение между аллелем е4 и болезнью дозозависимое; две копии е4 связаны с более ранним началом (средний возраст начала до 70 лет), чем одна копия (средний возраст начала после 70 лет). В отличие от этого, е2 аллель имеет защитный эффект и соответственно чаще встречается у пожилых, незатронутых болезнью Альцгеймера.
Механизмы, лежащие в основе данных эффектов, неизвестны, но полиморфные варианты АроЕ могут влиять на процессинг бета-АРР и плотность амилоидных отложений в мозге пациентов с болезнью Альцгеймера. Например, мыши без АроЕ имеют выраженное снижение депонирования пептида А-бета, получаемого из мутантного гена бета-АРР, связанного с семейной формой болезни Альцгеймера. Предполагают и другие механизмы, например измененный ответ на повреждение, так как ген АРОЕ управляется в мозге в процессе повреждения и восстановления. Важно иметь в виду, что е4 аллель АроЕ неоднозначно связан с повышенным риском болезни Альцгеймера. Таким образом, носители аллелей е4 имеют плохие неврологические результаты после черепно-мозговых травм, инсультов и других неврологических нарушений.
Хотя носители е4 аллеля АРОЕ имеют четко повышенный риск развития болезни Альцгеймера, к настоящему времени скрининг на присутствие этого аллеля у здоровых индивидуумов нецелесообразен; такое тестирование имеет высокие цифры ложноположительных и ложноотрицательных ответов и приводит к неопределенным оценкам риска болезни Альцгеймера.
Другие гены болезни Альцгеймера. Статистический анализ показывает, что риск болезни Альцгеймера могут значительно изменять еще 4-8 генов. Сущность их неясна. Кроме того, исследования типа случай-контроль при болезни Альцгеймера указывают на длинный список возможных генов (>100), но лишь несколько из них получили подтверждение при повторном анализе, и их роль в генетическом определении риска при болезни Альцгеймера остается неизвестной.
Видео этиология, патогенез болезни Альцгеймера (деменции)