• Ферменты репарации ДНК обеспечивают низкий уровень спонтанных мутаций
• Дефекты генов репарации ДНК увеличивают базальный уровень мутаций
• Мутации в генах, кодирующих белки точек проверки, приводят к нарушению целостности хромосом
Попытка рассчитать вероятность развития рака на основании известных данных о частоте мутагенеза и количестве мутантных генов, которые должны образоваться в геноме раковой клетки, представляет собой непростую задачу. Процесс канцерогенеза настолько многоступенчат, а скорость спонтанного мутагенеза столь мала, что практически невозможно понять, каким образом рак может развиться за 70-80 лет жизни человека.
Решение этого парадокса зависит от критического параметра, который включается в расчет. Это параметр частоты возникновения спонтанных мутаций. По-видимому, в расчете на поколение раковые клетки накапливают мутации с гораздо большей скоростью по сравнению со здоровыми клетками. Такая повышенная мутабильность сама по себе делает развитие рака математически вероятным.
Скорость спонтанного мутагенеза, с одной стороны, зависит от степени повреждений, которые различные канцерогены образуют в ДНК и скорости их копирования ДНК-полимеразой при репликации. С другой стороны, очевидно, что большинство ошибочных последовательностей, образующихся при повреждении ДНК или за счет ошибок копирования, быстро удаляются и заменяются правильными последовательностями.
Клетки обладают системой мониторинга, которая постоянно следит за целостностью генома, удаляет неправильные основания и заменяет их неповрежденными, восстанавливая первоначальную структуру ДНК. Эта система репарации ДНК эффективно удаляет основания, измененные за счет окислительных процессов и прочих повреждающих химических реакций. В целом, за счет системы репарации ДНК скорость образования мутаций снижается менее чем до 10-6 ген/поколение.
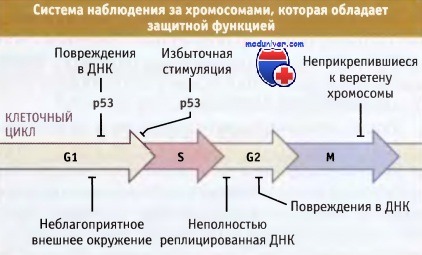
Наряду с этим, существуют «контрольные точки», которые гарантируют успешное завершение очередного критического этапа клеточного цикла до момента наступления следующего. Так, при повреждении задерживается синтез ДНК, митоз не начинается до момента завершения репликации, и блокируется в анафазе, при нарушении прикрепления хромосом к митотическому веретену.
Так же как и остальные внутриклеточные механизмы, о которых здесь шла речь, эти хранители генома могут повреждаться. Мутации в генах, кодирующих компоненты системы репарации ДНК, приводят к дефектной репарации и к повышению скорости образования мутантных аллелей. С уверенностью можно сказать, что эти мутации с одинаковой частотой возникают во всех участках генома, но среди поврежденных присутствует много генов, контролирующих рост клеток.
В клетке существует множество систем, которые постоянно следят за внешним окружением и за состоянием хромосом на протяжении клеточного цикла.
Эти системы могут замедлять или блокировать деление клеток до момента полной репарации повреждений.
В результате этого многие, если не все, этапы развития опухоли происходят более быстро. Вместо 5-10 лет каждый из них происходит в течение гораздо более короткого промежутка времени. Это приводит к тому, что многоэтапный процесс развития опухоли гораздо быстрее доходит до заключительного этапа, и пропорционально возрастает вероятность быстрого наступления клинических проявлений опухолевого процесса.
Критическую роль в остановке клеточного цикла после повреждения ДНК играет белок-супрессор опухолевого роста, р53. В ответ на многие типы физиологического стресса, р53 может на время заблокировать пролиферацию клеток или вызвать активацию программы апоптоза. Заболевание атаксия-телеангиэктазия представляет собой сложное патологическое состояние, которое в том числе приводит к развитию рака.
Заболевание передается по наследству с дефектным геном, кодирующим протеинкиназу, способствующую репарации ДНК от повреждений за счет фосфорилирования р53 и его активации. После активации р53 может индуцировать белок (называемый р21), который блокирует прохождение клеток по циклу. р53 может также инактивироваться за счет мутаций, затрагивающих хромосомный ген р53. Белок р53 может также инактивироваться при взаимодействии с онкобелком Е6 вируса папилломы человека.
Утрата белком р53 функциональной активности, наступающая по тому или иному механизму, спасает раковую клетку от постоянной опасности впасть в апоптоз, индуцированный белком р53.
Вместе с тем, в настоящее время известно много случаев семейных раков, при которых наследуемый ген кодирует тот или иной компонент сложного аппарата, ответственного за поддержание целостности генома. Первоначально обнаруженное заболевание такого типа называется пигментной ксеродермой (ХР). Больные ХР проявляют крайне высокую чувствительность к солнечному Уф. Это связано с тем, что они получили по наследству дефектные копии генов, кодирующих один из десяти или более белков, ответственных за обнаружение поврежденных УФ оснований ДНК, их выщепление и замещение неповрежденными основаниями. У таких больных солнечный свет вызывает сотни повреждений кожи, многие из которых развиваются в карциномы.
Хорошо известны гены BRCA1 и BRCA2, поскольку наследование дефектных аллелей предрасполагает к развитию карцином молочной железы и яичников. Эти гены кодируют белки, участвующие в репарации дву-нитевых разрывов ДНК. Заболевание наследственным неполипозным раком ободочной кишки (HNPCC) вызывается наследованием дефектных вариантов любого из четырех генов, участвующих в коррекции неспаренных оснований ДНК.
На рисунке ниже представлены различные системы внутриклеточного наблюдения, позволяющие защитить хромосомы от повреждений, а также мутации, ведущие к развитию рака.
Наконец, большинство раковых клеток человека являются анеуплоидными, которые в процессе малигнизации потеряли свой нормальный диплоидный кариотип. Существует точка зрения, согласно которой эта анеуплоидность обусловлена тем, что раковые клетки постоянно меняют карты своих хромосом; при этом могут образовываться части и дефектные плечи хромосом, которые благоприятствуют злокачественному росту.
Действительно, имеются данные, полученные на некоторых типах опухолей и свидетельствующие о том, что фактически при каждом злокачественном росте проявляется генетическая нестабильность или на уровне последовательностей ДНК, или на уровне кариотипа.
Такая генетическая нестабильность может представлять собой дефект, свойственный раковым клеткам, который играет столь же важную роль при малигнизации, как и дефекты, связанные с действием специфических генов-регуляторов роста.