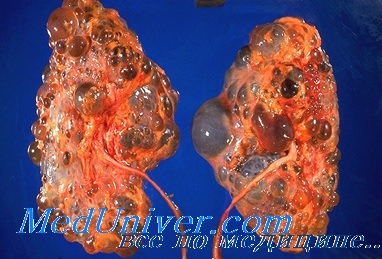
Поликистоз почек взрослых (ПКПВ). Артериопеченочная дисплазия - синдром Alagille
Поликистоз почек взрослых (ПКПВ) довольно распространенное аугосомио-доминантное заболевание, которым в США болеют около 500 тыс. чел.; в 8-10% случаев ПКПВ является основанием для назначения длительного гемодиализа. Развитие кист в почках зависит от возраста, поэтому попытка выявления гетерозигот до появления симптомов заболевания с помощью УЗИ может не дать результатов до наступления зрелого возраста.
Около 50% пациентов имеют ЛГ, 50% — кисты печени, у 50% и итоге развивается тяжелая почечная недостаточность, а у неопределенной (но, вероятно, высокой) части пациентов имеются дивертикулы толстой кишки. Повышенный уровень ренина в плазме крови участвует в развитии ЛГ задолго до наступления ПН. К нарушениям со стороны ССЗ относят: ПМК, который диагностируют в 25% случаев, умеренное расширение корня аорты, в редких случаях аневризмы грудной и брюшной аорты и предрасположенность к аортальной, митральной и трикуспидальной регургитации.
Аналогичное, но менее выраженное, чем при синдроме Marfan, сочетание дивертикулеза толстой кишки, кист в других органах и поражений ССС позволяет предположить вовлеченность в патогенез ПКПВ нарушений внеклеточного матрикса.
Наиболее серьезное проявление типичные мешотчатые аневризмы мозговых артерий (похожие на ягоду), которые развиваются у 10% гетерозигот и могут оставаться асимптоматичными в течение всей жизни. Наличие ЛГ предрасполагает к субарахноидальным кровоизлияниям. Вопрос о том, как диагностировать и лечить внутричерепные аневризмы у пациентов при отсутствии неврологических симптомов, остается спорным.
Повышенный риск проведения ангиографии сосудов головного мозга у больных ПКПВ связан с расслоением сосудистой стенки и усилением сосудистой реактивности. МРТ позволяет обнаружить большинство мешотчатых аневризм диаметром 2-3 мм. Возможность профилактического лечения небольших аневризм систематически не изучалась. Однако, несомненно, что всем пациентам с ПКПВ показан обязательный контроль артериального давления.
Причиной поликистоза почек взрослых являются нарушения но крайней мере в 3 генах. Большинство случаев связано с мутацией РВР в локусе гена PKD1. В остальных случаях наследственный ПКПВ связан с мутациями в локусе гена PKD2 на хромосоме 4 (4q23-q23). У пациентов из семей с мутацией в локусе гена PKD2 отмечается тенденция к более позднему развитию и более мягкому течению ПН.
В обоих случаях (PKD1 и PKD2), если в нормальном аллсле соответствующего мутантного локуса возникает соматическая мутация, у больных развиваются мультиорганные кисты по аналогии с неопластической трансформацией, которая происходит в соответствии с так называемой моделью «двух ударов» (two-hit model). (Согласно этой модели, опухоль развивается у индивидуума, гетерозиготного по мутантному гену, кодирующему доминантно-наследуемую форму заболевания, только при наличии дополнительной мутации в соматической клетке).
В одной из франко-канадских семей было выявлено заболевание, характерное для мутации гена PKD1, но не связанное ни с каким другим локусом; это послужило свидетельством существования гена PKD.1. Носители мутаций в генах PKD1 и PKD2 демонстрируют широкую внутрисемейную вариабельность. Обнаружение факторов, влияющих на эту вариабельность, позволит предложить новые подходы к контролю за прогрессированием болезни.
Артериопеченочная дисплазия
Высоковариабельное аутосомно-доминантное заболевание, известное как синдром Alagille, проявляется в виде желтухи новорожденных, вызванной недоразвитостью внутрииеченочных желчных протоков, а у наиболее тяжелобольных детей — застойной СН, хотя у гетерозиготных родственников заболевание может протекать бессимптомно.
Нарушения со стороны сердечно-сосудистой системы у большинства больных включают периферический стеноз легочной артерии и системных артерий, иногда связанный с дефектами перегородки или наличием открытого артериального протока (ОАП). В отдельных случаях отмечается диффузная васкулопатия. Патология почек может стать причиной развития АГ. Сначала локус был картирован с помощью хромосомного анализа, позволившего обнаружить у ряда больных частичную или полную утрату сегмента 20р12 (интерстициальная деления).
Установлено, что причиной заболевания в большинстве случаев служат мутации гена JAG1, кодирующего лиганд Notch-рецептора Jagged 1, который локализуется именно в этой области хромосомы. Впоследствии в ряде семей были обнаружены мутации гена NOTCH2.