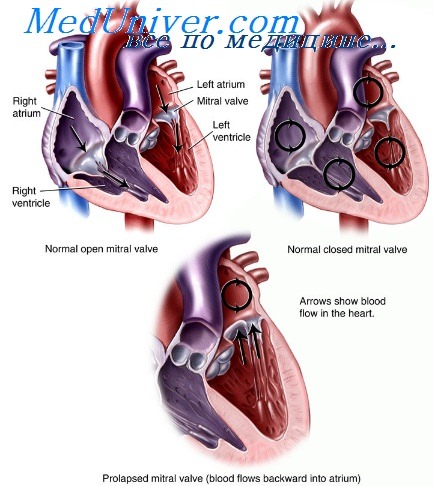
Пролапс митрального клапана при синдроме Marfan или Ehlers-Danlos. ПМК при синдроме истонченной дряблой кожи
Специалисты по медицинской генетике и врачи-кардиологи часто наблюдают пациентов с подозрением на синдром Marfan или Ehlers-Danlos. У некоторых таких больных нарушения не соответствуют минимальным диагностическим критериям известных заболеваний соединительной ткани, однако у больных имеются четкие экстракардиальные признаки, соответствующие повреждениям внеклеточного матрикса, которые будут описаны далее.
Часто, по не всегда обнаруживают пролапс митрального клапана; если у пациента имеется пролапс митрального клапана, а доказательства системного нарушения соединительной ткани отсутствуют, ему ставят диагноз «первичный ПМК». Спектр клинических симптомов при ПМК включает аномальные атрофические стрии, избыточный размах рук и длину ног, гипермобильность суставов, воронкообразную деформацию грудной клетки, сколиоз, уменьшение грудного кифоза («прямая спина»), миопию и незначительное расширение корня аорты.
Расширение аорты > 3 стандартных отклонений от средних значений, зависимых от величины поверхности тела, расслоение аорты, эктопия хрусталика или наличие любого из этих признаков в анамнезе позволяют исключить пациента из этой категории. Для остальных пациентов нарушения описываю!' как фенотип MASS, который являемся гетерогенной группировкой больных и семей. При этом особое внимание уделяют аорте, поскольку имеет место ее прогрессирующее расширение и расслоение; на самом деле ни одного такого случая не описано, т.к. проспективные наблюдения велись бессистемно.
Фенотипом MASS объясняют многие ассоциации между ПMK и деформацией грудной клетки и спонтанным пневмотораксом.
Наконец, пролапс митрального клапана часто сопровождает синдром Marfan, некоторые случаи синдрома Ehlers-Danlos и синдром истонченной дряблой кожи (cutis laxa); неожиданно часто ПМК обнаруживают у больных с несовершенным остеогенезом, синдромом Larsen, эластической псевдоксантомой и другими синдромами, наследуемыми по законам Менделя. В отдельных семьях встречаются не классифицированные дисплазии соединительной ткани, характеризующиеся существенными нарушениями аппарата митрального клапана с миксоматозной дегенерацией, кальнификацией или с тем и другим.
Обнаружено, что некоторые пациенты и члены их семей, имеющие признаки синдрома Marfan, по не удовлетворяющие строгим критериям диагностики, имеют мутации в гене, колирующем рецептор 1 типа трансформирующего фактора роста В (TGFBR1, transforming growth factor-beta receptor-1). Позднее развитие у членов семьи аневризмы аорты, наследуемой только аутосомно-доминантно, также стали связывать с мутациями этого гена. У пациентов с различными фенотипами, включая тех, у кого в течение многих лет диагностировали неполный синдром Marfan, Loeys B.L. и соавт. обнаружили мутации в генах, кодирующих как TGFBR, так и TGFBR2.
Среди наиболее тяжелых фенотипических проявлений у пациентов отмечают краниосипостоз, широко расставленные глаза (гипертелоризм), удвоение небного язычка или расщелину неба, косолапость, арахнодактилию, эктазию твердой мозговой оболочки, открытый артериальный проток, расширение восходящей аорты, извитость аорты и выраженную предрасположенность к расслоению аорты даже при ее минимальном расширении. У этих пациентов не развивается эктопия хрусталика, они имеют невысокий рост. Для подтверждения диагноза важно провести молекулярно-генетический анализ и выявить родственников, унаследовавших мутацию.
Эффективным оказывается профилактическое восстановление корня аорты, что показано даже при меньших размерах корня аорты, чем у больных с синдромом Marfan (т.е. < 45 мм для взрослых). Эффективность В-АБ или БРА не изучалась.