Кардиомиопатии - кратко с точки зрения внутренних болезней
Кардиомиопатии представляют собой первичное поражение миокарда и классифицируются по структурно-функциональному принципу (рис. 1). Кардиомиопатии могут иметь наследственный характер или быть связанными с инфекциями или воздействием токсинов.
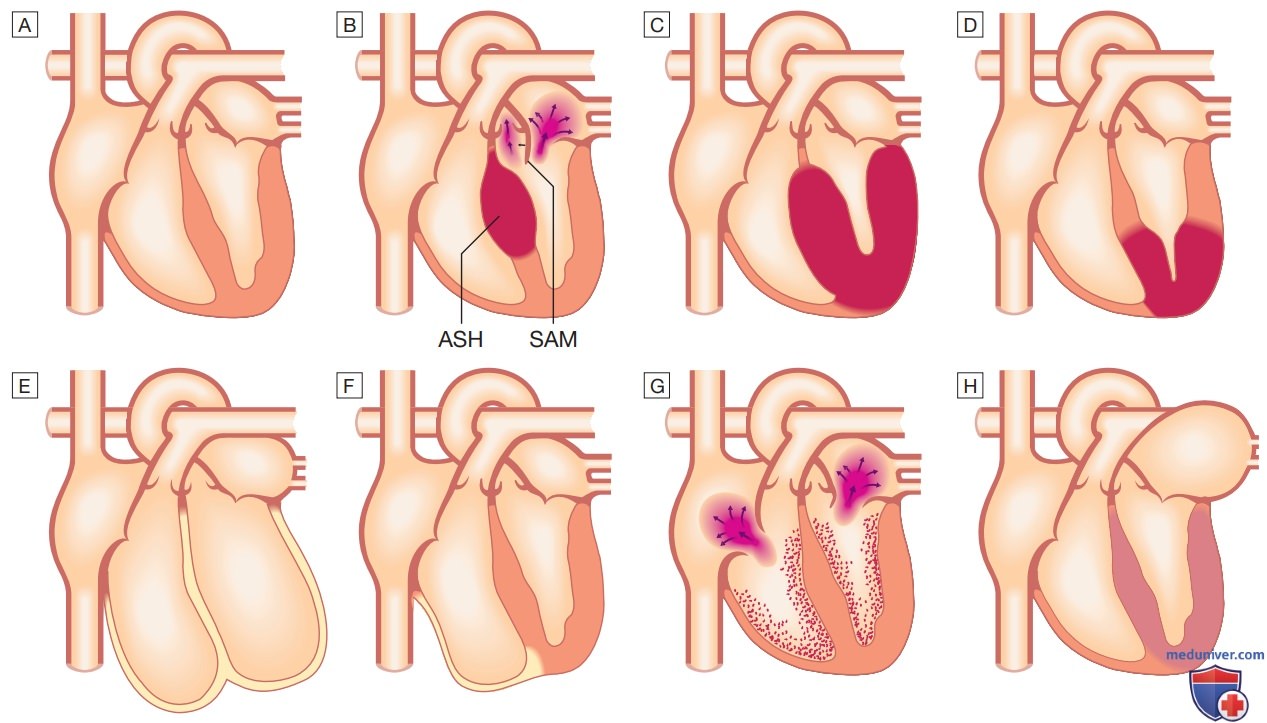
Рисунок 1. Типы кардиомиопатии: А — нормальное сердце; B — гипертрофическая кардиомиопатия: асимметричная гипертрофия межжелудочковой перегородки (Asymmetric Septal Hypertrophy — ASH) с передним систолическим движением митрального клапана (Systolic Anterior Motion — SAM), вызывающим митральную регургитацию и динамическую обструкцию выносящего тракта левого желудочка; C — гипертрофическая кардиомиопатия: концентрическая гипертрофия; D — гипертрофическая кардиомиопатия: гипертрофия верхушки сердца; E — дилатационная кардиомиопатия; F — аритмогенная кардиомиопатия правого желудочка; G — облитерирующая кардиомиопатия (в общепринятых классификациях рассматривается как эндомиокардиальный вариант рестриктивной кардиомиопатии с эозинофилией или без нее); H — рестриктивная кардиомиопатия
а) Дилатационная кардиомиопатия. В Северной Америке и Европе заболеваемость дилатационной кардиомиопатией с клиническими проявлениями составляет 20 на 100 000 человек, а распространенность — 38 на 100 000 человек. Мужчины болеют более чем в 2 раза чаще, чем женщины.
1. Патогенез. Кардиомиопатия характеризуется дилатацией ЛЖ и зачастую ПЖ с нарушением их сократимости. Масса ЛЖ увеличена, однако толщина стенки нормальная или снижена (рис. 1). Расширение фиброзных колец клапанов сердца может повлечь за собой функциональную митральную и трикуспидальную недостаточность. Гистологические изменения могут быть различными, включают повреждения миофибрилл, интерстициальный фиброз и инфильтраты, состоящие из Т-лимфоцитов.
Термин «дилатационная кардиомиопатия» охватывает гетерогенную группу заболеваний. У некоторых пациентов значимой причиной может быть злоупотребление алкоголем. По крайней мере в 25% случаев дилатационная кардиомиопатия наследуется по аутосомно-доминантному типу, были выявлены различные мутации единичных генов. Большинство из этих мутаций затрагивают белки цитоскелета миоцитов, такие как дистрофин, ламины А и С, эмерин и метавинкулин. Многие из них также связаны с аномалиями скелетных мышц.
С другой стороны, большинство скелетных мышечных дистрофий, для которых характерно Х-сцепленное наследование, такие как миодистрофия Беккера и Дюшенна, сопровождаются развитием кардиомиопатии. Наконец, считается, что у значительной части пациентов с дилатационной кардиомиопатией причиной заболевания является поздняя аутоиммунная реакция после перенесенного вирусного миокардита. Полагают, что с подобным механизмом также связано поражение миокарда, которое встречается у 10% пациентов с ВИЧ-инфекцией.
2. Клиническая картина. Большинство пациентов обращаются за медицинской помощью в связи с симптомами сердечной недостаточности, либо заболевание у них обнаруживают случайно во время планового обследования. Нарушения ритма, тромбоэмболические осложнения и внезапная смерть могут возникать на любой стадии кардиомиопатии, наиболее частым симптомом являются эпизоды болей в грудной клетке. Дифференциальная диагностика включает нарушение функции желудочков вследствие ИБС, диагноз дилатационной кардиомиопатии ставят при исключении ишемического генеза.
3. Лабораторные и инструментальные исследования. Наибольшей диагностической ценностью обладают эхокардиография и МРТ сердца. Изменения на ЭКГ, хотя и возникают достаточно часто, являются неспецифическими. Генетическое исследование показано в случаях, когда заболевание выявлено более чем у одного члена семьи.
4. Лечение. Целью лечения является контроль за сердечной недостаточностью, который был описан выше. Несмотря на то что у некоторых пациентов в течение многих лет симптомы отсутствуют, прогноз может быть различным, и некоторым больным показана трансплантация сердца. Пациенты с дилатационной кардиомиопатией и среднетяжелой или тяжелой сердечной недостаточностью подвержены риску внезапной аритмической смерти, который можно снизить с помощью медикаментозной терапии β-адреноблокаторами и ингибиторами АПФ или БРА. У некоторых пациентов может быть рассмотрен вопрос об имплантации кардиовертера-дефибриллятора и/или устройства для СРТ.
б) Гипертрофическая кардиомиопатия. Гипертрофическая кардиомиопатия является наиболее частой формой кардиомиопатии, заболеваемость ею составляет приблизительно 100 на 100 000 человек. Она характеризуется нефизиологичной сложной гипертрофией ЛЖ с нарушением упорядоченности волокон миокарда и развитием фиброза. Гипертрофия может быть генерализованной, ограничиваться преимущественно межжелудочковой перегородкой (асимметричная гипертрофия межжелудочковой перегородки) (рис. 1) или другими участками сердца. Существует особый вариант в виде апикальной гипертрофической кардиомиопатии, который распространен на Дальнем Востоке.
1. Патогенез. Гипертрофическая кардиомиопатия — генетическое заболевание, которое обычно наследуется по аутосомно-доминантному типу с высокой степенью пенетрантности и вариабельной экспрессией генов. У большинства пациентов гипертрофическая кардиомиопатия обусловлена точечной мутацией в одном из генов, кодирующих сократительные белки саркомера. Существуют три общие группы мутаций с разными фенотипами. Мутации тяжелой цепи β-миозина приводят к развитию сложной гипертрофии желудочков. Мутации тропонина связаны с небольшой гипертрофией, в некоторых случаях она даже может отсутствовать, однако отмечаются выраженное разупорядочивание волокон миокарда, артериальная гипотензия при физических нагрузках и высокий риск внезапной смерти.
Мутации в гене миозинсвязывающего белка С, как правило, проявляются в позднем возрасте и часто ассоциированы с АГ и нарушениями ритма. При всех подтипах гипертрофической кардиомиопатии может развиваться сердечная недостаточность, поскольку диастолическое наполнение жесткого, неподатливого ЛЖ нарушено. Гипертрофия межжелудочковой перегородки может также приводить к динамической обструкции выходного тракта ЛЖ (гипертрофическая обструктивная Кардиомиопатия) и митральной недостаточности, связанной с передним систолическим движением передней створки митрального клапана.
2. Клиническая картина. К основным клиническим проявлениям относят симптомы, возникающие при физической нагрузке, в частности стенокардию, одышку, нарушения ритма и внезапную смерть. Симптомы и признаки гипертрофической кардиомиопатии сходны с таковыми при аортальном стенозе, за исключением скачущего пульса при гипертрофической кардиомиопатии (табл. 105). Годовая смертность, обусловленная внезапной смертью, составляет 2—3% среди взрослых и 4—6% — среди детей и подростков (табл. 106). Внезапная смерть, как правило, происходит во время или сразу после интенсивной физической нагрузки, причиной ее считаются желудочковые нарушения ритма сердца.
Гипертрофическая кардиомиопатия является наиболее частой причиной внезапной смерти у молодых спортсменов. У пациентов, у которых нет фатальных нарушений ритма, естественное течение болезни может быть различным, однако часто происходит медленное клиническое ухудшение.
3. Лабораторные и инструментальные исследования. Эхокардиографическое исследование является методом выбора и обычно позволяет поставить диагноз. В некоторых случаях диагностика бывает затруднительной, в частности, при наличии другой причины гипертрофии ЛЖ, однако степень гипертрофии при гипертрофической кардиомиопатии обычно больше, чем при физиологической гипертрофии, поражение чаще носит асимметричный характер. На ЭКГ обнаруживаются изменения, характерные для гипертрофии ЛЖ, с разнообразными, часто необычными нарушениями, включая глубокие отрицательные зубцы Т.
Может быть выполнено генетическое исследование, которое также позволяет провести скрининг родственников пациента.
4. Лечение. β-Адреноблокаторы, недегидропиридиновые антагонисты кальция и дизопирамид позволяют облегчить симптомы и предотвратить синкопальные состояния. Нарушения ритма часто поддаются терапии амиодароном. В тоже время фармакологического лечения, которое было бы способно улучшать прогноз, не существует. Обструкцию выносящего тракта ЛЖ можно устранить с помощью частичной хирургической резекции (миомэктомии) или путем создания ятрогенного инфаркта базальных отделов межжелудочковой перегородки с введением этилового спирта через катетер (спиртовая септальная абляция). У пациентов с клиническими факторами риска внезапной смерти следует рассмотреть вопрос об имплантации ИКД (табл. 106).
Дигоксин и вазодилататоры могут увеличить обструкцию выносящего тракта ЛЖ, поэтому их применения следует избегать.
в) Аритмогенная кардиомиопатия правого желудочка. При аритмогенной кардиомиопатии ПЖ наблюдается преимущественное поражение миокарда ПЖ. Заболевание наследуется по аутосомно-доминантному типу, заболеваемость приблизительно составляет 10 на 100 000 человек. Генетический дефект затрагивает гены белков десмосом, чаще всего плакофилина 2 (РКР2); тем не менее при использовании современных протоколов генетического исследования ген, виновный в развитии заболевания, во многих случаях выявить не удается. Аритмогенная кардиомиопатия ПЖ характеризуется заменой участков миокарда ПЖ фиброзной и жировой тканью (см. рис. 1). В некоторых случаях также отмечается поражение ЛЖ, при этом прогноз заболевания хуже.
Диагноз основан на комплексе критериев, учитывающих данные ЭКГ, структурные особенности, генетические изменения и нарушения ритма сердца.
Наиболее значимой клинической проблемой являются желудочковые нарушения ритма, внезапная смерть и правожелудочковая сердечная недостаточность. На ЭКГ, как правило, определяется незначительное расширение комплекса QRS и инверсия зубцов Т в правых пре-кардиальных отведениях. Большую диагностическую ценность имеет МРТ, которая наряду с ЭКГ в 12 отведениях и суточным мониторированием ЭКГ используется при скрининге родственников пациента первого порядка. Лечение направлено на коррекцию правожелудочковой недостаточности с помощью диуретиков и нарушений ритма сердца путем назначения β-адреноблокаторов. Пациентам с высоким риском внезапной смерти может быть предложена имплантация кардиовертера-дефибриллятора.
г) Рестриктивная кардиомиопатия. При данном редком заболевании наполнение желудочков нарушается вследствие их жесткости (см. рис. 1). Это приводит к повышению давления в предсердиях и их гипертрофии, дилатации и в дальнейшем к ФП. В Великобритании наиболее частой причиной рестриктивной кардиомиопатии является амилоидоз, хотя могут встречаться и другие виды поражения, например, при гликогенозах, идиопатическом перимиоцитарном фиброзе и при семейных формах рестриктивной кардиомиопатии. Диагностика заболевания может быть затруднительной и требует проведения допплер-эхокардиографии, КТ или МРТ и эндомиокардиальной биопсии. Лечение симптоматическое, но прогноз обычно плохой, некоторым пациентам может потребоваться трансплантация сердца.
д) Облитерирующая кардиомиопатия. Облитерирующая кардиомиопатия представляет собой редкую форму рестриктивной кардиомиопатии с вовлечением эндокарда одного или обоих желудочков. Данный вид кардиомиопатии характеризуется формированием тромбоза и фиброза с постепенной облитерацией полостей желудочков фиброзной тканью (см. рис. 1). Возникает недостаточность митрального и трикуспидального клапанов. Характерно развитие сердечной недостаточности, ТЭЛА и системных эмболий. Иногда облитерирующая кардиомиопатия ассоциирована с эозинофилией, кроме того, она может возникать при эозинофильном лейкозе и эозинофильном гранулематозе с полиангиитом (ранее известном как синдром Черга—Стросс). В тропических странах с заболеванием может быть связано до 10% случаев сердечной смерти. Смертность при облитерирующей кардиомиопатии высокая и составляет 50% в течение 2 лет.
В лечении используются антикоагулянтная и антиагрегантная терапия, для уменьшения симптомов сердечной недостаточности могут быть назначены диуретики. В отдельных случаях эффективно хирургическое лечение (протезирование трикуспидального и/или митрального клапана с удалением эндокарда).
е) Кардиомиопатия такоцубо. Кардиомиопатия такоцубо (синдром такоцубо) представляет собой вариант острой дисфункции ЛЖ, характеризующийся дилатацией верхушки ЛЖ и прилегающего к ней миокарда в сочетании с поражением ЛЖ. Механизм данного синдрома изучен недостаточно, он может включать норадренергическую вазоконстрикцию коронарных артерий и острую обструкцию выносящего тракта ЛЖ. Синдром такоцубо часто связан с острым стрессом, обусловленным внешним воздействием, или эмоциональным стрессом (например, потеря близкого человека) и сопровождается болью в грудной клетке, одышкой и иногда сердечной недостаточностью. Синдром такоцубо чаще развивается у женщин, чем у мужчин. И по симптоматике, и по изменениям на ЭКГ заболевание может напоминать острый коронарный синдром с подъемом сегмента ST.
Диагноз обычно ставится на основании коронаро-ангиографии при минимальных признаках ИБС или их отсутствии. При эхокардиографии определяется характерное баллонирование верхушки ЛЖ. Расширенная верхушка и узкий выносящий тракт ЛЖ по форме напоминают японскую ловушку для осьминогов, которая называется такоцубо (рис. 2). Функция ЛЖ обычно восстанавливается в течение 4—5 дней, хотя в некоторых случаях дисфункция может сохраняться на протяжении нескольких недель. Для лечения используются β-адреноблокаторы в целях профилактики аритмий и ингибиторы АПФ для лечения дисфункции ЛЖ. Такая терапия продолжается только до восстановления функции сердца.
Рисунок 2. Кардиомиопатия такоцубо. Вентрикулограмма левого желудочка после введения контраста у пациента с кардиомиопатией такоцубо. Контур расширенного левого желудочка указан стрелками
ж) Вторичные причины кардиомиопатии. При многих системных заболеваниях, включая заболевания соединительной ткани, саркоидоз, гемохроматоз и алкогольное поражение сердца, клиническая картина будет неотличима от таковой при дилатационной кардиомиопатии (табл. 107). В противоположность этому при амилоидозе и эозинофильной болезни сердца возникают симптомы и признаки, сходные с симптомами рестриктивной или облитерирующей кардиомиопатии. Поражение сердца при атаксии Фридрейха может быть похожим на гипертрофическую кардиомиопатию.
Лечение и прогноз зависят от основного заболевания. У пациентов с алкогольным поражением миокарда отказ от приема алкоголя в ряде случаев приводит к быстрому и значимому улучшению.